Nuevos artículos
Nuevos hallazgos contribuyen a comprender mejor las causas del síndrome de Rett
Último revisado: 02.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.

El síndrome de Rett es un trastorno poco común del neurodesarrollo para el cual actualmente no existe cura ni tratamiento eficaz. Provoca síntomas físicos y cognitivos graves, muchos de los cuales se solapan con los trastornos del espectro autista.
El síndrome de Rett está causado por mutaciones en el gen MECP2, que se expresa considerablemente en el cerebro y parece desempeñar un papel importante en el mantenimiento de la salud neuronal. Este gen se encuentra en el cromosoma X y el síndrome afecta principalmente a niñas. Para desarrollar tratamientos para el síndrome de Rett, los investigadores buscan comprender mejor el gen MECP2 y sus funciones en el cerebro.
Investigadores, entre ellos Rudolf Jaenisch, cofundador del Instituto Whitehead, han estudiado MECP2 durante décadas; sin embargo, muchos datos básicos sobre el gen aún se desconocían. La proteína codificada por el gen, MECP2, participa en la regulación génica; se une al ADN e influye en los niveles de expresión de varios otros genes, o en la cantidad de proteína que producen.
Sin embargo, los investigadores no tenían una lista completa de genes afectados por MECP2 y no había consenso sobre cómo MECP2 afecta a estos genes.
Estudios preliminares de MECP2 sugirieron que era un represor, reduciendo la expresión de sus genes diana, pero investigaciones de Jaenisch y otros habían demostrado previamente que MECP2 también actúa como activador, aumentando la expresión de sus genes diana, y que podría ser un activador en primer lugar. También se desconocía el mecanismo de acción de MECP2, o qué hace exactamente la proteína para provocar cambios en la expresión génica.
Las limitaciones tecnológicas han impedido a los investigadores aclarar estas preguntas. Sin embargo, Yanish, el investigador posdoctoral de su laboratorio, Yi Liu, y el exmiembro del laboratorio de Yanish, Anthony Flamier, ahora profesor adjunto en el centro de investigación CHU Sainte-Justine de la Universidad de Montreal, han utilizado técnicas de vanguardia para responder a estas preguntas pendientes sobre MECP2 y obtener nuevos conocimientos sobre su papel en la salud y las enfermedades cerebrales.
Sus resultados fueron publicados en la revista Neuron y los investigadores también crearon un repositorio en línea de sus datos MECP2, el portal MECP2-NeuroAtlas, como recurso para otros investigadores.
"Creo que este artículo cambiará radicalmente la comprensión de cómo MECP2 causa el síndrome de Rett. Tenemos una comprensión completamente nueva del mecanismo y podría brindar nuevas vías para el desarrollo de tratamientos para la enfermedad", afirma Janisch, quien también es profesor de biología en el MIT.
Una comprensión más profunda de MECP2 en el cerebro
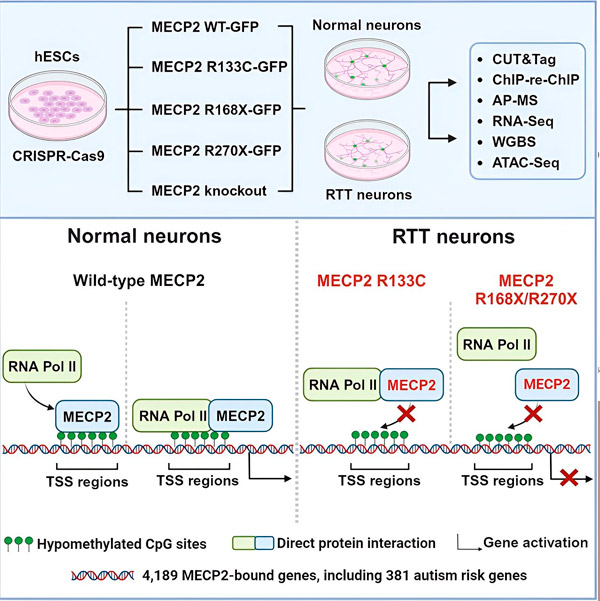
Los investigadores primero crearon un mapa detallado de la unión de MECP2 en las secuencias génicas neuronales humanas, ya sea dentro de los genes o en las regiones reguladoras del ADN cercanas. Utilizaron un método llamado CUT&Tag, que permite identificar con gran precisión las interacciones de las proteínas con el ADN.
Los investigadores encontraron más de 4000 genes asociados con MECP2. Repitieron su mapeo en neuronas con mutaciones comunes de MECP2 asociadas con el síndrome de Rett para determinar dónde se encuentra MECP2 disminuido durante la enfermedad.
Saber a qué genes se une MECP2 permitió a Liu y Flamier comenzar a establecer conexiones entre los objetivos de MECP2 y la salud cerebral. Descubrieron que muchos de sus objetivos participan en el desarrollo y la función de los axones y las sinapsis neuronales.
También compararon su lista de objetivos de MECP2 con la base de datos de genes asociados al autismo de la Iniciativa de Investigación del Autismo de la Fundación Simons (SFARI) y encontraron que 381 genes en esa base de datos son objetivos de MECP2.
Fuente: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Estos hallazgos pueden ayudar a aclarar los mecanismos subyacentes a los síntomas del autismo en el síndrome de Rett y proporcionar un buen punto de partida para investigar el posible papel de MECP2 en el autismo.
"Hemos creado el primer mapa integrado del epigenoma de MECP2 en la salud y la enfermedad, y este mapa puede guiar la investigación futura", afirma Liu. "Saber qué genes son diana de MECP2 y cuáles se ven directamente afectados en la enfermedad proporciona una base sólida para comprender el síndrome de Rett y plantear preguntas sobre la regulación génica neuronal".
Los investigadores también analizaron si MECP2 aumentaba o disminuía la expresión de sus genes diana. En consonancia con la historia de que algunos identifican MECP2 como activador y otros como represor, Liu y Flamier encontraron ejemplos donde MECP2 desempeñaba ambas funciones.
Sin embargo, aunque MECP2 se considera más a menudo un represor, Liu y Flamier descubrieron que es principalmente un activador, lo que confirma hallazgos previos de Jaenisch y Liu. Un nuevo experimento demostró que MECP2 activa al menos el 80 % de sus dianas, y otro descubrió que activa hasta el 88 % de sus dianas.
El mapa de genes diana creado por los investigadores proporcionó información adicional sobre la función de MECP2 como activador. Descubrieron que, en el caso de los genes que MECP2 activa, este se une típicamente a una región de ADN aguas arriba del gen, denominada sitio de inicio de la transcripción.
Este es el sitio donde la maquinaria celular inicia el proceso de transcripción de un gen a ARN, tras lo cual este se traduce a una proteína funcional, producto de la expresión génica. La presencia de MECP2 en el sitio de inicio de la transcripción, donde comienza la expresión génica, concuerda con su función como activador génico.
Los investigadores se propusieron determinar el papel de MECP2 en la activación génica. Analizaron a qué moléculas se une MECP2 en este sitio, además del ADN, y descubrieron que MECP2 interactúa directamente con un complejo proteico llamado ARN polimerasa II (ARN Pol II). La ARN Pol II es una máquina celular clave que transcribe el ADN en ARN. La ARN Pol II no puede encontrar genes por sí sola, por lo que requiere diversos cofactores, o proteínas colaboradoras, para realizar su función.
Los investigadores proponen que MECP2 actúa como uno de estos cofactores, ayudando a la ARN Pol II a iniciar la transcripción en los genes a los que se une. El análisis estructural de MECP2 ha identificado partes de la molécula que se unen a la ARN Pol II, y otros experimentos han confirmado que la pérdida de MECP2 reduce la presencia de la ARN Pol II en los sitios de inicio de la transcripción apropiados, así como los niveles de expresión de los genes diana.
Esto sugiere que el síndrome de Rett podría deberse a una disminución de la transcripción de los genes diana de MECP2 debido a mutaciones de MECP2 que impiden su unión a la ARN Pol II o al ADN. En consonancia con esta idea, las mutaciones de MECP2 más comunes asociadas con la enfermedad son truncamientos: mutaciones en las que falta parte de la proteína, lo que puede alterar la interacción entre MECP2 y la ARN Pol II.
Los investigadores esperan que sus hallazgos no sólo cambien nuestra comprensión de MECP2, sino que una comprensión más profunda y amplia de cómo MECP2 influye en el desarrollo y la función del cerebro podría conducir a nuevos conocimientos que ayudarán a las personas con síndrome de Rett y trastornos relacionados, incluido el autismo.
"Este proyecto es un excelente ejemplo del carácter colaborativo del laboratorio de Janisch", afirma Flamier. "Rudolf y yo teníamos un problema específico relacionado con el síndrome de Rett, y yo tenía experiencia con la tecnología CUT&Tag, que podría resolverlo. Tras conversar, nos dimos cuenta de que podíamos aunar esfuerzos, y ahora contamos con un amplio repositorio de información sobre MECP2 y su relación con la enfermedad".