Médico experto del artículo.

Nuevos artículos
Atrofia muscular espinal
Último revisado: 29.06.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
La atrofia muscular espinal no es una unidad nosológica única, sino un grupo completo de patologías hereditarias clínica y genéticamente heterogéneas, provocadas por los procesos crecientes de degeneración de las motoneuronas de las astas espinales anteriores. El término abarca diferentes variantes de paresia periférica y atrofia muscular genéticamente determinadas, resultantes de la degeneración de las motoneuronas espinales o del tronco encefálico. La causa más común del problema es una mutación autosómica recesiva en el hombro q largo del quinto cromosoma. El tratamiento es inespecífico y se centra en mejorar la troficidad del tejido nervioso y proporcionar apoyo paliativo para mejorar la calidad de vida. [ 1 ]
Epidemiología
La atrofia muscular espinal ocurre en un caso por cada 6.000 a 10.000 recién nacidos (según el American Journal of Medical Genetics 2002).
La prevalencia de portadores de deleción del exón 7 del gen SMN es de 1:50 personas.
La atrofia muscular bulboespinal (síndrome de Kennedy) se presenta en un niño de cada 50.000 y es el tipo más común de amiotrofia espinal en adultos.
Se observa que la mitad de los niños con esta enfermedad no superan el período de supervivencia de dos años.
La patología se hereda según el principio autosómico recesivo. Con frecuencia, cada progenitor de un niño enfermo es portador de una copia del gen mutado. Dado que la mutación se compensa con la presencia de una segunda copia "normal" del gen, los progenitores no presentan manifestaciones de atrofia muscular espinal. La patología de tipo 2 no suele heredar una copia adicional del progenitor. El problema se produce debido a un fallo accidental durante la formación de las células germinales o directamente en el momento de la fecundación. En la atrofia muscular espinal del primer tipo, el desarrollo espontáneo de la enfermedad ocurre solo en el 2% de los casos (en este caso, solo uno de los progenitores es portador). [ 2 ]
Causas atrofia muscular espinal
La principal causa de la atrofia muscular espinal es una mutación del gen responsable de la producción de la proteína SMN, localizado en el cromosoma 5q. Este trastorno provoca además la muerte gradual de las células nerviosas motoras en las astas anteriores de la médula espinal y el tronco encefálico. Como resultado de estos procesos, el tono muscular disminuye y se desarrolla atrofia de los músculos respiratorios, faríngeos, faciales y esqueléticos. El tipo de herencia predominante en las formas pediátricas de atrofia muscular espinal es autosómico recesivo, lo que implica la portación simultánea de genes defectuosos por parte de ambos progenitores. En cuanto a la patología de tipo IV (forma adulta), existe un vínculo con el cromosoma X, por lo que solo los varones se ven afectados.
El desarrollo de la atrofia muscular espinal se basa en la creciente degeneración y muerte de las neuronas motoras de las astas anteriores espinales y el daño a los núcleos del tronco encefálico. Los cambios patológicos son más intensos en las zonas de engrosamiento cervical y lumbar. El número de células se reduce al mínimo y se produce su reemplazo por tejido conectivo, lo que se debe a un fallo en el programa de muerte celular, la llamada apoptosis. El cambio afecta las estructuras de los núcleos motores de los nervios craneales, las raíces anteriores y los nervios motores. Existe un cuadro clínico de atrofia fascicular neurogénica. Con un curso prolongado de la enfermedad, en una etapa tardía, se produce un sobrecrecimiento del tejido conectivo.
La aparición del cuadro clínico correspondiente se asocia a una deficiencia de la proteína SMN, que afecta el correcto funcionamiento de las células nerviosas motoras en las astas espinales anteriores. La deficiencia de proteína, como uno de los factores que contribuyen al desarrollo de la atrofia muscular espinal, se descubrió a finales del siglo XX. En el contexto del daño de las motoneuronas, se altera la inervación de los músculos esqueléticos (principalmente las secciones proximales). [ 3 ]
Factores de riesgo
La diversidad de formas clínicas de la atrofia muscular espinal 5q se explica por la presencia de ciertos factores modificadores que pueden dividirse en dos categorías: los que afectan y los que no afectan el puntaje de la proteína SMN.
- Actualmente, el gen SMN2 se considera el factor fundamental en el desarrollo de la atrofia muscular espinal: cuantas más copias del gen SMN2, menor es la intensidad de los síntomas de la enfermedad. El segundo factor, directamente relacionado con la copia centromérica del gen SMN, es una sustitución de un nucleótido c.859G>C en el exón 7 del gen SMN2, que conduce a la formación de un nuevo sitio de empalme de unión al potenciador: el resultado es la inclusión del exón 7 en la transcripción del gen SMN2. Esta variación se asocia con un aumento en la concentración sanguínea de la proteína SMN completa en pacientes con amiotrofia espinal de segundo o tercer tipo.
Otros factores que afectan el número de SMN:
- Factores reguladores del empalme (Tra2β - induce la omisión del exón 7, SF2/ASF - aumenta la inclusión del exón 7, hnRNPA1 - suprime la inclusión del exón 7 del gen SMN2).
- Factores reguladores de la transcripción (CREB1 - aumenta la transcripción de SMN, STAT3 - favorece el crecimiento axonal, IRF1 - aumenta el número de SMN, PRL - aumenta la esperanza de vida en estadios graves).
- Factores estabilizadores del ARNm (U1A reduce SMN, HuR/p38).
- Factores que afectan la modificación postraduccional (RCA: suprime la degradación de SMN, GSK3: aumenta la supervivencia).
- Factores exógenos (inanición, hipoxia, estrés oxidativo).
Los efectos de los factores mencionados anteriormente se determinaron predominantemente in vitro.
- Factores que no están asociados con el gen SMN, en particular, proteínas que optimizan la endocitosis en las sinapsis (laminina 3, coronina, neurocalcina delta, proteína similar a la neurina de calcio).
Se presta especial atención a la metilación del ADN, la modificación más estable que afecta la naturaleza de la expresión génica. Se ha descubierto que la metilación de un grupo de genes posiblemente involucrados en procesos patogénicos se correlaciona con la gravedad de la atrofia muscular espinal. [ 4 ]
Patogenesia
La atrofia muscular espinal es una patología genética con herencia autosómica dominante, autosómica recesiva o ligada al cromosoma X. Generalmente, se trata de una patología autosómica recesiva en la primera infancia. El gen SMN, localizado en el locus 5q13, es responsable de la formación de esta amiotrofia espinal. La deleción del exón 7 del gen SMN provoca una patología con posible afectación de los genes p44 y NAIP, ambos adyacentes.
El genoma del SNM codifica una proteína de 294 aminoácidos con una masa molecular de aproximadamente 38 kDa. Esta proteína cumple las siguientes funciones:
- Es parte del complejo ARN-proteína;
- Participa en la formación del sitio del espliceosoma que cataliza el empalme del pre-ARN;
- Participa en los procesos que controlan la producción de proteínas y las isoformas de proteínas;
- Proporciona transporte axonal de ARNm;
- Favorece el crecimiento de las células nerviosas y proporciona comunicación neuromuscular.
Se conocen un par de tipos de genes SMN:
- SMNt telomérico (SMN1);
- SMNc centromérico (SMN2).
La gran mayoría de los casos de atrofia muscular espinal se deben a alteraciones en el gen SMN1.
La atrofia muscular espinal de Kennedy está ligada al locus Xq12, que contiene el gen NR3C3, que codifica una proteína del receptor de andrógenos. Presenta una variante de herencia ligada al cromosoma X. Cuando aumenta el número de repeticiones de CAG en el exón de un gen, se desarrolla la patología.
La supresión de la producción de proteína SNM se acompaña de los siguientes cambios:
- Debido a una coordinación axonal deteriorada, se produce una ramificación excesiva de los axones;
- El crecimiento de los axones se ralentiza y su tamaño disminuye;
- Hay una agrupación inadecuada de los canales de calcio en el cono de crecimiento;
- Se forman terminales presimpáticas irregulares de los axones de las células nerviosas motoras.
La médula espinal comienza a perder activamente neuronas motoras en los cuernos anteriores, lo que explica el desarrollo de la atrofia de los músculos proximales de las extremidades. [ 5 ]
Síntomas atrofia muscular espinal
La sintomatología de la atrofia muscular espinal de Werdnig-Hoffman se presenta con mayor frecuencia en el período neonatal y hasta los seis meses, manifestándose por el síndrome del bebé perezoso. Se observan tórax acampanado, hipotonía intensa, ausencia de reflejos, espasmos musculares en la lengua y dificultad respiratoria. Los bebés enfermos fallecen con mayor frecuencia antes de los dos años: el desenlace fatal se debe al aumento de la insuficiencia respiratoria en el contexto de la persistencia de procesos infecciosos.
La forma intermedia de atrofia muscular espinal de segundo tipo se detecta a partir de los seis meses de edad. Además del síndrome del niño "perezoso", se presentan hipotensión, falta de reflejos, trastornos respiratorios y tics linguales. Incluso si los niños pueden sentarse, se desarrollan múltiples contracturas en las grandes articulaciones.
La atrofia muscular espinal de Kugelberg-Wielander también comienza en la primera infancia, cuando los niños pueden moverse de forma independiente. Se presenta debilitamiento de los músculos ilíaco, cuádriceps y aductor, hipotensión, disminución de los reflejos y espasmos linguales. Muchos pacientes pierden la capacidad de moverse (caminar) de forma independiente con el paso de los años.
La atrofia muscular espinal tipo 4 comienza a una edad más avanzada. Se caracteriza por una progresión lenta y un pronóstico relativamente benigno. [ 6 ]
La atrofia de Kennedy se manifiesta con mayor frecuencia en la mediana edad (generalmente puede debutar en pacientes de 15 a 60 años). La sintomatología incluye dolor y debilidad muscular, ginecomastia, debilidad distal, letargo, tics linguales y atrofia. Los signos de disfunción bulbar son:
- Dificultad para tragar;
- Aspiración;
- Debilitamiento de los músculos masticatorios;
- Disartria;
- Temblores posturales y motores en las manos.
Primeros signos de deficiencia de andrógenos:
- Ginecomastia (en aproximadamente el 60% de los pacientes), a menudo asimétrica;
- Deterioro de la función sexual (oligospermia, atrofia testicular, disfunción eréctil).
Primeros signos
La amiotrofia espinal se manifiesta por debilidad muscular e impotencia generalizada. No se ven afectadas las capacidades sensoriales ni intelectuales.
Principales índices de patología neuromuscular:
- Se nota musculatura "perezosa", debilitada, flacidez y laxitud de los músculos;
- El tono muscular es bajo, los reflejos tendinosos están minimizados o ausentes;
- Reflejos plantares normales o ausentes;
- Se observan contracciones cortas de grupos musculares individuales (se pueden ver debajo de la piel, en la lengua);
- Hay signos de atrofia muscular.
El síndrome de Werdnig-Hoffman se manifiesta por hipotonía muscular pronunciada, letargo general e incapacidad del niño para sostener la cabeza, darse la vuelta y sentarse. Al intentar sostener al bebé en el abdomen en estado de suspensión, el cuerpo parece "hundirse". Los reflejos de tos, deglución y succión son insatisfactorios, el alimento suele penetrar en las vías respiratorias y la respiración es problemática. Puede haber distorsión articular asociada con la hipotonía intrauterina. La información anamnésica obtenida durante el embarazo suele indicar baja actividad fetal.
Signos básicos de la atrofia muscular espinal tipo I:
- Retraso severo en el desarrollo motor;
- Aparición rápida de contracturas articulares y curvatura torácica;
- Aumento de trastornos respiratorios y bulbares, problemas con la deglución (tanto de alimentos como de saliva) y expectoración de esputo;
- Mayor riesgo de inflamación por aspiración;
- Infección, insuficiencia respiratoria progresiva.
La atrofia muscular espinal tipo II se manifiesta por una clara inhibición del desarrollo motor. Aunque muchos pacientes pueden sentarse sin ayuda, e incluso a veces gatear y ponerse de pie, estas capacidades suelen perderse con el tiempo. Se observan temblores en los dedos, distorsiones musculares y articulares (óseas), y problemas respiratorios. Posible pseudohipertrofia de la pantorrilla.
Las principales características de la patología tipo II:
- Retrasos en el desarrollo, incluida la detención y reversión del desarrollo de habilidades y capacidades ya adquiridas;
- Debilidad creciente de los músculos intercostales;
- Superficialidad de la respiración diafragmática, reflejo de la tos debilitado, empeoramiento gradual de la insuficiencia respiratoria;
- Curvatura del tórax y columna vertebral, contracturas.
En el síndrome de Kugelberg-Wielander, las manifestaciones son más leves y progresan lentamente. El paciente puede desplazarse, pero presenta dificultades para correr o subir escaleras. Los síntomas tardíos suelen incluir dificultad para tragar y masticar.
La atrofia muscular espinal tipo IV se manifiesta ya en la edad adulta y se caracteriza por una evolución leve y favorable. Los principales signos son la pérdida gradual de la capacidad de movimiento. [ 7 ]
Formas
La atrofia muscular espinal forma parte de un grupo de patologías hereditarias caracterizadas por cambios degenerativos, la muerte de las células nerviosas motoras de las astas espinales anteriores y, a menudo, de los núcleos motores del tronco encefálico. El proceso puede manifestarse en diferentes etapas de la vida, y el cuadro clínico no siempre es el mismo. Los tipos de herencia y la evolución también pueden variar.
La atrofia muscular espinal pediátrica se describió por primera vez a finales del siglo XIX. A mediados del siglo XX, se identificaron las principales formas de la enfermedad:
- Congénita (se manifiesta casi inmediatamente después del nacimiento del bebé);
- Forma infantil temprana (se produce en el contexto de un desarrollo normal previo del bebé);
- Forma infantil tardía (se manifiesta a partir de los 2 años de edad y más).
Algunos especialistas combinan la segunda y la tercera forma en un tipo pediátrico de amiotrofia espinal.
Generalmente se acepta dividir la patología en pediátrica y adulta. La atrofia muscular espinal en niños se clasifica en temprana (con inicio en los primeros meses tras el nacimiento), tardía y adolescente (juvenil). Los síndromes más comunes son:
- Atrofia de Werdnig-Hoffman;
- La forma Kugelberg-Wielander;
- Atrofia muscular espinal infantil crónica;
- Síndrome de Vialetto-van Lare (tipo bulboespinal con ausencia de audición);
- Síndrome de Fazio-Londe.
La atrofia muscular espinal en adultos se presenta entre los 16 y los 60 años aproximadamente, y se caracteriza por una clínica y un pronóstico relativamente benignos. Las patologías en adultos incluyen:
- Atrofia bulboespinal de Kennedy;
- Atrofia escapuloperoneal;
- Formas facial-lapez-hombro y óculo-faríngea;
- Atrofia espinal distal;
- Atrofia espinal monomélica.
Se distinguen la atrofia espinal aislada y la combinada. La patología aislada se caracteriza por el predominio de daño en las neuronas motoras espinales (que a menudo es el único signo del problema). La patología combinada es poco frecuente y representa un complejo de trastornos neurológicos y somáticos. Se han descrito casos de síndrome combinado con malformaciones coronarias congénitas, pérdida de la función auditiva, oligofrenia e hipoplasia cerebelosa.
La atrofia muscular espinal en personas mayores se manifiesta con mayor frecuencia por la amiotrofia bulboespinal de Kennedy. Esta patología se hereda de forma recesiva ligada al cromosoma X. La enfermedad tiene una evolución lenta y relativamente benigna. Comienza con atrofia de la musculatura proximal de las extremidades inferiores. Es posible que se presente temblor en las manos y la cabeza. Simultáneamente, también se detectan problemas endocrinos: atrofia testicular, ginecomastia y diabetes mellitus. A pesar de ello, en adultos, la patología es más leve que en niños.
Una variante de la atrofia muscular espinal. |
El debut de la patología |
Problema detectable |
Edad de muerte |
Sintomatología característica |
Atrofia muscular espinal tipo 1 (otro nombre: atrofia muscular espinal de Verding-Hoffman) |
Desde el nacimiento hasta los seis meses |
El bebé no puede sentarse |
Hasta dos años |
Debilidad muscular grave, hipotonía, dificultad para mantener la cabeza erguida, alteración del llanto y la tos, problemas para tragar y salivar, desarrollo de insuficiencia respiratoria y neumonía por aspiración. |
Atrofia muscular espinal tipo 2 |
De seis meses a un año y medio |
El bebé no puede mantenerse en pie |
Más de dos años |
Retraso motor, deficiencia de peso, debilidad por tos, temblores en las manos, curvatura de la columna, contracturas. |
Atrofia muscular espinal tipo 3 (otro nombre: atrofia muscular espinal de Kugelberg-Welander) |
Después de un año y medio. |
Al principio puede estar de pie y caminar, pero a cierta edad esta capacidad puede perderse. |
En la edad adulta. |
Músculos debilitados, contracturas, hipermovilidad articular. |
Atrofia muscular espinal tipo 4. |
Adolescencia o adultez |
Al principio puede estar de pie y caminar, pero a cierta edad esta capacidad puede perderse. |
En la edad adulta. |
Aumento de la debilidad muscular proximal, disminución de los reflejos tendinosos, espasmos musculares (fasciculaciones). |
La atrofia espinal distal se asocia con lesiones en las células nerviosas motoras de la médula espinal, que inervan la parte inferior del cuerpo. Los signos característicos de esta patología son:
- Atrofia de los músculos del muslo;
- Debilidad en las rodillas, extensores del pie y músculos aductores de la cadera.
No hay cambios en los reflejos tendinosos.
La atrofia muscular espinal distal está representada por dos variaciones alélicas con un fenotipo superpuesto:
- Atrofia muscular espinal escápulo-perineal;
- Neuropatía sensitivo-motora hereditaria de Charcot-Marie-Tooth tipo 2C.
La atrofia muscular espinal proximal 5q se caracteriza por una sintomatología creciente de parálisis flácida y atrofia muscular, debida a cambios degenerativos en las motoneuronas alfa de las astas espinales anteriores. La enfermedad congénita con asfixia posparto es la forma más grave: desde el nacimiento, la actividad motora es prácticamente nula, presenta contracturas y problemas de deglución y respiratorios. En la mayoría de los casos, el niño fallece.
Complicaciones y consecuencias
La progresión de la amiotrofia espinal provoca debilidad y reducción de la masa muscular en las extremidades (especialmente en las piernas). Inicialmente, el bebé no posee las habilidades adquiridas o las pierde gradualmente; es decir, pierde la capacidad de caminar y sentarse sin apoyo. La actividad motora de las extremidades superiores disminuye, las articulaciones se vuelven rígidas, con el tiempo se forman contracturas y la columna vertebral se curva.
Para preservar las capacidades motoras durante el mayor tiempo posible y prevenir el desarrollo de complicaciones, se recomienda:
- Practique una postura corporal correcta (posición antigravedad), tanto en la cama como al sentarse, caminar, etc.;
- Fisioterapia regular, ejercicios de estiramiento, masajes, fisioterapia, independientemente del tipo de atrofia muscular espinal;
- Utilice camas, sillas (sillas de ruedas), colchones y almohadas especiales;
- Seleccionar y utilizar aparatos ortopédicos y corsés de apoyo;
- Practicar hidroterapia y kinesioterapia, que tienen un efecto favorable sobre el aparato respiratorio, musculoesquelético y digestivo, el sistema nervioso y cardiovascular;
- Realizar controles diagnósticos periódicos, incluyendo exámenes clínicos, radiografías de columna y pelvis;
- Consultar sistemáticamente con un fisioterapeuta y un ortopedista con experiencia en el trabajo con pacientes similares;
- Ajustar corsés, órtesis, aparatos ortopédicos, sillas de ruedas, etc. según la dinámica.
Los cuidadores de un paciente con atrofia muscular espinal deben estar familiarizados con:
- Con los fundamentos de conducta segura, fisioterapia, masajes, fisioterapia;
- Con las reglas de mantenimiento de la actividad independiente del paciente, uso de dispositivos ortopédicos;
- Con las normas de cuidado, higiene.
La amiotrofia espinal suele complicarse por alteraciones en la masticación, la deglución y la conducción de los alimentos, lo que amenaza la aspiración y el desarrollo de inflamación pulmonar por aspiración u obstrucción de las vías respiratorias, características más comunes de la patología del primer tipo. Los problemas de deglución se manifiestan mediante síntomas como prolongación significativa y persistente del tiempo para comer, reticencia a comer, caída de la comida, náuseas frecuentes y empeoramiento de la pérdida de peso.
Los trastornos de la motilidad digestiva se manifiestan como estreñimiento, peristaltismo débil, retención gástrica prolongada (estasis gástrica) y reflujo gastroesofágico. Para prevenir estas complicaciones es necesario:
- Vigilar la posición correcta del paciente al comer;
- Si es necesario, utilice una sonda gástrica o una gastrostomía para asegurar una ingesta adecuada de líquidos y nutrientes y reducir el riesgo de aspiración;
- Respetar las normas de preparación de alimentos y bebidas, vigilar su consistencia y la frecuencia de las comidas;
- Dependiendo de la prescripción médica, utilizar medicación, masajes, fisioterapia, etc.
Una de las complicaciones más graves de la amiotrofia espinal es la disfunción del sistema respiratorio asociada con la debilidad de los músculos respiratorios. Los trastornos respiratorios pueden ser mortales, tanto en bebés con patología tipo 1 como en adolescentes y adultos con enfermedad tipo 2 o 3. Los problemas clave son los siguientes:
- El reflejo de la tos se altera, hay problemas con la expectoración de esputo del tracto respiratorio;
- Aumento del déficit del volumen de aire que entra a los pulmones, alteración de la excreción de dióxido de carbono de los pulmones;
- Distorsiona el pecho, comprime y deforma los pulmones;
- Procesos infecciosos en forma de bronconeumonía.
Para prevenir tales complicaciones, a menudo se recomienda a los pacientes realizar ejercicios de respiración utilizando una bolsa Ambu. [ 9 ]
Diagnostico atrofia muscular espinal
En pacientes con sospecha de amiotrofia espinal, investigaciones como las siguientes tienen valor diagnóstico:
- Química sanguínea;
- Análisis genético de ADN;
- Electroneuromiografía.
Entre los métodos adicionales se pueden citar la biopsia de las fibras musculares, la ecografía y la resonancia de los músculos y del cerebro.
Los análisis de sangre pueden indicar que la creatina fosfoquinasa es fisiológicamente normal, pero en algunos casos puede estar elevada hasta aproximadamente 2,5 veces.
El electroneuromiograma revela cambios debidos a la pérdida de neuronas motoras espinales. Esto se detecta por una disminución en la amplitud de la curva de interferencia y la aparición de potenciales activos espontáneos, que son fibrilaciones y fascioculaciones que forman un ritmo de frecuencia específico. La velocidad de la señal de impulso que pasa a través de las fibras motoras periféricas es normal o está disminuida debido a trastornos secundarios de denervación. [ 10 ]
El diagnóstico instrumental suele realizarse también mediante ecografía o resonancia magnética de la musculatura, que permite detectar la sustitución muscular por tejido graso. La resonancia magnética revela un patrón patológico característico de la atrofia muscular espinal. Sin embargo, esto solo es posible en las etapas avanzadas de la lesión.
Durante el análisis morfológico de la biopsia muscular en pacientes, se detecta un cuadro inespecífico, caracterizado por atrofia de haces musculares y agrupamiento de fibras musculares. La gran mayoría de las fibras musculares afectadas pertenecen al tipo 1, y las características inmunohistológicas y químicas se encuentran dentro de los límites normales. El cuadro ultraestructural es inespecífico.
El procedimiento diagnóstico más importante ante la sospecha de atrofia muscular espinal es la prueba que detecta la mutación del gen SMN. Mediante el análisis directo del ADN, es posible detectar la presencia o ausencia de los exones séptimo y octavo de los genes SMNc y SMNt. El método más informativo es el análisis cuantitativo, que permite determinar el número de copias del gen y determinar la forma de la atrofia muscular espinal. El método cuantitativo también es importante para evaluar el estado del paciente. Es una medida necesaria para el asesoramiento familiar médico y genético.
Se realizan pruebas diagnósticas adicionales solo tras obtener un resultado negativo de la deleción del gen SMN. Si se requiere la detección de mutaciones puntuales, se puede utilizar la secuenciación automatizada directa del gen SMNt.
Diagnóstico diferencial
El diagnóstico diferencial se realiza con procesos patológicos que presentan el complejo sintomático de un paciente perezoso, con distrofias musculares congénitas y miopatía estructural o mitocondrial. En particular, debe descartarse la presencia de estas patologías:
- Enfermedad de la neurona motora;
- Miosclerosis lateral primaria;
- Distrofia muscular;
- Miopatías congénitas;
- Enfermedades asociadas con la acumulación de glucógeno;
- Polio;
- Miastenia gravis autoinmune.
El algoritmo diagnóstico se desarrolla en función de las peculiaridades de la sintomatología de cada niño. Por lo tanto, se utiliza una clasificación específica de pacientes según su estado funcional (Europrotocolo TREAT-NMD):
- Incapaz de sentarse sin apoyo (postrado en cama).
- Capaz de sentarse pero no puede caminar (sedentario).
- Capaz de moverse de forma independiente (pacientes que caminan).
Para los pacientes del primer grupo se recomienda el siguiente algoritmo diagnóstico:
- Examen físico (detección de la curvatura del tórax, evaluación de la función respiratoria y de la tos, y del estado de la piel);
- Monitorización cardíaca y respiratoria, polisomnografía e identificación de síntomas de déficit de ventilación pulmonar;
- Oximetría de pulso para determinar el grado de oxigenación;
- Evaluación de la frecuencia de patologías infecciosas-inflamatorias y cursos de antibióticos durante el semestre extremo;
- Radiografías de tórax con estudios de dinámica repetida;
- Evaluación de la función deglutoria.
Para los pacientes del segundo grupo, se aplica el siguiente algoritmo:
- Examen físico;
- Monitorización cardíaca y respiratoria, polisomnografía para detectar déficit de ventilación pulmonar;
- Oximetría de pulso;
- Evaluación de la frecuencia de procesos infecciosos-inflamatorios y cursos de antibióticos durante el período extremo de seis meses;
- Examen de la columna vertebral, radiografías de la columna, evaluación del grado de curvatura.
Los pacientes del tercer grupo están indicados para dichos estudios:
- Examen físico;
- Pruebas de la función respiratoria (incluye espirometría, cálculo del volumen pulmonar, evaluación de la función de los músculos respiratorios);
- Conocer la frecuencia de patologías infecciosas-inflamatorias y cursos de antibióticos durante el período anual extremo.
El diagnóstico diferencial puede complicarse por la similitud de los genes SMN1 y SMN2. Para evitar errores, se recomienda utilizar el método MLPA, que permite detectar el número de copias del exón 7 del gen SMN1.
En la mayoría de los casos de atrofia muscular espinal, se observa una deleción homocigótica del exón 7 y/u 8 del gen SMN1. Sin embargo, otros genes (ATP7A, DCTN1, UBA1, BSCL2, EXOSC3, GARS, etc.) también pueden ser responsables, por lo que se debe prestar atención si la prueba de SMN1 es negativa.
El biomaterial para el estudio puede ser sangre periférica o fetal, o mapas de gotas de sangre seca. El diagnóstico es obligatorio:
- En presencia de antecedentes agravados de atrofia muscular espinal;
- Cuando se detectan síntomas sospechosos, independientemente de los antecedentes hereditarios.
Además, también se recomienda la investigación a todas las parejas que son responsables de planificar un embarazo.
¿A quién contactar?
Tratamiento atrofia muscular espinal
Los pacientes con atrofia muscular espinal necesitan un tratamiento integral que incluya:
- Cuidado, ayuda, apoyo;
- Comida dietetica;
- Terapia farmacológica;
- Medidas de rehabilitación sin medicamentos, incluida la kinesioterapia y la fisioterapia.
Un régimen terapéutico que implica un efecto polimodal en todos los sistemas del cuerpo, no sólo en el sistema musculoesquelético, es estándar.
Desafortunadamente, es imposible curar radicalmente la atrofia muscular espinal. Sin embargo, a menudo es posible mejorar la calidad de vida del paciente mediante el uso adecuado de aminoácidos y complejos multivitamínicos, agentes neurotróficos, bloqueadores de los canales de calcio, vasodilatadores, fármacos cardiotróficos y citostáticos, inhibidores de la proteasa, esteroides, antioxidantes, inmunoglobulinas e inmunosupresores, entre otros. Se ha demostrado experimentalmente que el tratamiento con células madre, compuestos neuroprotectores y moléculas de fortalecimiento muscular puede provocar trastornos sistémicos impredecibles. Sin embargo, hasta la fecha no se han demostrado efectos positivos tras la aplicación de dicho tratamiento.
Dado que el problema se debe a una deficiencia de la proteína SMN normal, los pacientes pueden mejorar aumentando los niveles de proteína SMN en un 25 % o más. Por ello, se están investigando activamente fármacos que pueden activar la producción de esta proteína, como la gabapentina, el riluzol, la hidroxiurea, el albuterol, el ácido valproico y el fenilbutirato de sodio.
La medicina moderna también ofrece tratamiento quirúrgico para la atrofia muscular espinal. Consiste en la alineación quirúrgica de la columna vertebral y la corrección de la curvatura neuromuscular. Los cirujanos realizan una fijación multinivel de la columna vertebral mediante estructuras especiales. El sacro, la pelvis y las vértebras torácicas superiores, entre otras, se utilizan como puntos de apoyo. La cirugía ayuda a alinear la columna vertebral, distribuir uniformemente la carga sobre ella, eliminar las molestias al cambiar de posición y evitar efectos adversos en los órganos internos (incluidos los pulmones).
Medicamentos
Actualmente, no existe un tratamiento etiológico para la atrofia muscular espinal: la medicina científica continúa trabajando en esta tarea. Anteriormente, los científicos ya han logrado aislar fármacos que pueden aumentar la producción de ARNm del gen SMN2. Sin embargo, aún no se han realizado ensayos clínicos internacionales a gran escala con personas con atrofia muscular espinal.
La mayoría de los medicamentos incluidos en el régimen de tratamiento estándar tienen un principio de acción general con evidencia de eficacia relativamente baja.
L-Carnitina |
Un aminoácido natural, pariente de las vitaminas del grupo B. Se produce en el organismo, está presente en el hígado y el músculo estriado transverso, y pertenece a varias sustancias similares a las vitaminas. Participa en los procesos metabólicos, favorece la actividad de la CoA y se utiliza para normalizar el metabolismo. Tiene propiedades anabólicas, antitiroideas y antihipóxicas, estimula el metabolismo lipídico y la reparación tisular, y optimiza el apetito. La L-carnitina se prescribe en una dosis de aproximadamente 1000 mg al día. El tratamiento puede durar hasta dos meses. |
Coenzima Q10 (ubiquinona) |
Una coenzima del grupo benzoquinona que contiene varios grupos isoprenilo. Estas coenzimas son liposolubles y se encuentran principalmente en las mitocondrias de las estructuras celulares eucariotas. La ubiquinona forma parte de la cadena de transporte de electrones y participa en la fosforilación oxidativa. Su mayor presencia se encuentra en órganos ricos en energía, en particular en el hígado y el corazón. Entre otras cosas, la coenzima Q10 posee propiedades antioxidantes y puede restaurar la capacidad antioxidante del alfa-tocoferol. Generalmente se prescriben de 30 a 90 mg del fármaco al día, durante un tratamiento de dos meses. |
Cerebrolisina |
Un fármaco nootrópico con propiedades neurotróficas. Se utiliza frecuentemente en regímenes terapéuticos para el tratamiento de patologías neurológicas, como la demencia vascular y el ictus. Su fracción activa incluye péptidos con un peso molecular límite de 10 mil daltons. El fármaco se administra mediante inyección intravenosa de 1 a 2 ml. El tratamiento consta de 10 a 15 inyecciones. |
Actovegin |
La composición del fármaco está compuesta por péptidos de bajo peso molecular y derivados de aminoácidos. Actovegin es un hemoderivado: se aísla mediante diálisis con ultrafiltración. Gracias a su uso, se aumenta la absorción y utilización del oxígeno, y se acelera el metabolismo energético. El fármaco se administra en inyecciones intravenosas de 1 a 2 ml; el tratamiento requiere de 10 a 15 inyecciones. |
Solcoseril |
Es un hemodializado desproteinizado capaz de optimizar el transporte precelular de oxígeno y glucosa, potenciar la producción intracelular de ATP, estimular las reacciones regenerativas tisulares, activar la proliferación de fibroblastos y la producción de colágeno en las paredes vasculares. El tratamiento consiste en 10-15 inyecciones intramusculares del fármaco (1-2 ml al día). |
Neuromultivit (complejo de vitamina B) |
Multivitamínico de uso activo para la deficiencia de vitaminas del grupo B. Con frecuencia, puede ser un sustituto eficaz de las inyecciones de preparados vitamínicos. Activa los procesos metabólicos en el cerebro, promueve la regeneración de los tejidos del sistema nervioso y tiene un efecto analgésico. Neuromultivit se toma de 1 a 2 comprimidos al día durante 4 u 8 semanas. |
Vitamina E |
Una vitamina liposoluble y antioxidante muy conocida. Se prescribe en tratamientos de 1 a 2 meses en dosis de 10 a 20 UI diarias. |
Valproato |
Tienen actividad sedante y relajante, poseen capacidad anticonvulsiva y aumentan los niveles de GABA en el sistema nervioso central (SNC). Se utilizan solo para el tratamiento de niños mayores de un año, en dosis de 10 a 20 mg por kg al día. |
Salbutamol |
Un broncodilatador que pertenece al grupo de los agonistas selectivos de los receptores beta2-adrenérgicos. Su uso regular aumenta la producción de ARNm y proteína SMN, lo que mejora el cuadro clínico de la atrofia muscular espinal. El salbutamol se usa con precaución, en dosis de 2 a 4 mg cuatro veces al día (la dosis máxima es de 32 mg al día). |
Uno de los fármacos más recientes utilizados para la atrofia muscular espinal es el genoterapéutico Zolgensma, que garantiza la actividad y el correcto funcionamiento de las células nerviosas motoras transducidas. Este fármaco se administra en combinación con inmunomoduladores según un protocolo especial y se administra una sola vez por vía intravenosa, a una dosis nominal de 1,1 × 10¹⁻¹ Vg/kg (el volumen total administrado se determina en función del peso del paciente).
Antes de iniciar el tratamiento con Zolgensma, es fundamental determinar el nivel de anticuerpos contra AAV9 mediante un método diagnóstico validado, evaluar la función hepática (ALT, AST, bilirrubina total), realizar un análisis de sangre clínico general y una prueba de troponina I, y determinar la creatinina. Si se detectan infecciones activas, tanto agudas como crónicas, la administración del fármaco se pospone hasta la curación o la finalización de la fase de recaída del proceso infeccioso.
El efecto secundario más frecuente del medicamento se considera la insuficiencia hepática, que puede ser mortal.
Otros medicamentos aprobados que su médico puede recetarle para la atrofia muscular espinal:
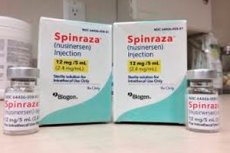
- Spinraza es una preparación de nusinersén sódico, un oligonucleótido antisentido diseñado específicamente para el tratamiento de la amiotrofia espinal. Se administra por vía intratecal mediante punción lumbar. La dosis recomendada es de 12 mg. El régimen de tratamiento lo determina el médico tratante.
- Risdiplam es un fármaco que modifica el empalme del ARNm precursor del gen 2 de supervivencia de las células nerviosas motoras. Risdiplam se administra por vía oral una vez al día. La dosis la determina el médico individualmente, teniendo en cuenta la edad y el peso del paciente. Está contraindicado su uso en niños menores de 2 meses. Se ha observado toxicidad embriofetal, por lo que las pacientes con potencial reproductivo deben tomar precauciones anticonceptivas durante el tratamiento y durante un período posterior.
Tratamiento fisioterapéutico de la atrofia muscular espinal
La fisioterapia se utiliza como uno de los componentes de la terapia compleja y la rehabilitación de pacientes con atrofia muscular espinal. Los principales puntos de este tratamiento son:
- Utilización de descarga mediante sistemas de suspensión, entrenamiento activo-pasivo, utilización de estimulación eléctrica percutánea de la médula espinal;
- Ejercicios de respiración y fisioterapia;
- Sesiones de verticalización de media hora;
- Tratamientos de electroestimulación translingual (sesiones de 20 minutos, combinadas con ejercicios para mejorar la motricidad fina);
- Técnicas manuales;
- Aplicaciones de parafina en diferentes grupos de articulaciones;
- Darsonval para mejorar el rendimiento muscular.
El método de darsonvalización se basa en el efecto sobre los tejidos mediante una corriente pulsada alterna de alta frecuencia, de alto voltaje y baja intensidad. Tras un ciclo de procedimientos, se observa un aumento del rendimiento muscular, el fortalecimiento de la microcirculación, la expansión de arteriolas y capilares, la eliminación de la isquemia y la mejora de la nutrición y el suministro de oxígeno a los músculos, lo que repercute positivamente en el curso de los procesos regenerativos y atróficos.
Uno de los problemas más importantes en pacientes con amiotrofia espinal es la debilidad de los músculos respiratorios, que a menudo conduce a disfunción respiratoria y a la muerte del paciente.
En la amiotrofia espinal, toda la musculatura esquelética, incluida la responsable de la respiración, presenta un rendimiento deficiente. La debilidad y la atrofia muscular gradual afectan negativamente la calidad de la respiración, provocando complicaciones y un aumento de la insuficiencia respiratoria. Por lo tanto, es necesario tomar medidas para fortalecer la musculatura y prevenir complicaciones e infecciones respiratorias. La gimnasia con la bolsa Ambu desempeña un papel especial en este sentido, junto con fisioterapia, ejercicios de estiramiento y masajes. El uso de la bolsa Ambu permite expandir el volumen del tórax y los pulmones. Para las actividades infantiles, es adecuada una bolsa con un volumen mínimo de un litro y medio, equipada con una válvula para liberar el exceso de presión (para prevenir el barotrauma).
Los ejercicios no deben realizarse con el estómago lleno. Posición corporal: sentado, semisentado, acostado de lado o boca arriba (si no hay problemas de flema): lo ideal es realizar los procedimientos en diferentes posiciones cada vez. Es importante que el paciente tenga la espalda recta. Si es necesario, se utiliza un corsé. Antes de comenzar el procedimiento, asegúrese de que las vías respiratorias estén libres de flemas.
Masaje para la atrofia muscular espinal
El masaje para el tratamiento de la amiotrofia espinal debe ser ligero y suave. En las zonas de resistencia muscular, aplicar efectos generales, como golpeteos, y en las zonas de inervación preservada, masajes profundos (longitudinales, transversales) y amasamientos.
En general, se practican diferentes tipos de masaje, según las características individuales de la enfermedad y la edad del paciente. Estos pueden ser:
- Amasamiento para estimular los músculos profundos;
- Frotaciones para optimizar la circulación sanguínea y linfática;
- Tratamiento localizado de puntos gatillo;
- Del golpeteo fortalecedor de las fibras.
Es importante que el efecto se distribuya en toda el área problemática.
Contraindicaciones del masaje para la atrofia muscular espinal:
- Inflamación aguda, temperatura corporal elevada;
- Trastornos sanguíneos, tendencias hemorrágicas;
- Procesos purulentos;
- Enfermedades dermatológicas infecciosas y fúngicas;
- Aneurismas vasculares, tromboflebitis, endarteritis, linfadenitis;
- Neoplasias benignas y malignas.
El tratamiento de cualquier masaje para un paciente con atrofia muscular espinal se prescribe de forma estrictamente individualizada. Una ejecución incorrecta del procedimiento, un impacto excesivamente brusco o incorrecto, puede perjudicar la condición del paciente.
Prevención
Actualmente se está trabajando activamente en el diagnóstico directo e indirecto de ADN, así como en el diagnóstico prenatal de ADN. Esto reduce significativamente la probabilidad de que nazca un bebé enfermo, lo cual es especialmente importante para las parejas que ya han tenido hijos con atrofia muscular espinal.
Las medidas preventivas representan una tendencia médica importante y se clasifican en medidas primarias, secundarias y terciarias.
Las medidas primarias tienen como objetivo prevenir directamente la influencia de un factor desfavorable y prevenir el desarrollo de la enfermedad. Dicha prevención consiste en corregir la dieta y la rutina diaria, y llevar un estilo de vida saludable.
La prevención secundaria consiste en la eliminación de los factores de riesgo evidentes e incluye el diagnóstico precoz de las patologías, el establecimiento de una vigilancia en dinámica y el tratamiento dirigido.
La prevención terciaria se aplica a personas enfermas con discapacidad motora. En este caso, se trata de rehabilitación farmacológica, psicológica, social y laboral.
Según información de la Organización Mundial de la Salud, más del 2% de los bebés en el mundo nacen con algún tipo de trastorno del desarrollo. Al mismo tiempo, entre el 0,5% y el 1% de estos trastornos son de origen genético. La prevención de estos problemas se reduce al asesoramiento genético médico y a un diagnóstico prenatal de calidad, lo que permite minimizar los riesgos de dar a luz a un bebé con patología genética.
El riesgo de una persona de padecer atrofia muscular espinal u otra enfermedad genética depende de los genes heredados de su madre y su padre. La identificación temprana de factores hereditarios y el cálculo del riesgo individual de padecer una patología genética pueden considerarse una forma de prevención específica.
Las medidas de diagnóstico prenatal incluyen métodos de investigación directos e indirectos. Inicialmente, se identifica a las mujeres que requieren diagnóstico prenatal indirecto. Estos pueden incluir:
- Mujeres embarazadas de 35 años de edad o más;
- Que hayan tenido 2 o más abortos espontáneos previos;
- Que tengan hijos con defectos genéticos del desarrollo;
- Con una historia hereditaria desfavorable;
- Que hayan tenido infecciones virales o exposición a la radiación (incluso durante la etapa de planificación del embarazo).
Con fines preventivos, se utilizan métodos como la ecografía y las pruebas hormonales (cribado bioquímico). En ocasiones, también se emplean procedimientos invasivos como la coriobiopsia, la amniocentesis, la placentocentesis y la cordocentesis. La información fiable sobre los riesgos genéticos permite adaptar el estilo de vida y el embarazo para prevenir el nacimiento de un niño enfermo.
Vacuna contra la atrofia muscular espinal
Por supuesto, a todos los padres de niños con amiotrofia espinal les gustaría curarlos por completo. Sin embargo, no existe una vacuna que pueda erradicar el problema. Aunque se están realizando investigaciones para optimizar el tratamiento.
En particular, en 2016, los científicos estadounidenses aprobaron el fármaco único Spinraza (nusinersen), que posteriormente fue aprobado para su uso en países europeos.
Los especialistas están investigando el problema del tratamiento de la atrofia muscular espinal de estas maneras:
- Reparar o reemplazar el gen SMN1 "incorrecto";
- Potenciación de la función del gen SMN2 normal;
- Protección de las células nerviosas motoras afectadas por la deficiencia de la proteína SMN;
- Protección de los músculos frente a cambios atróficos para prevenir o restaurar la función perdida en el contexto del desarrollo de la patología.
La terapia génica consiste en atacar el gen dañado mediante vectores virales que atraviesan la membrana hematoencefálica y alcanzan la zona correspondiente de la médula espinal. Posteriormente, el virus infecta la célula afectada con una porción sana de ADN, como si se tratara de una sutura del gen defectuoso. De esta manera, se corrige la función de las células nerviosas motoras.
Otra área de estudio es la terapia de moléculas pequeñas, cuyo objetivo principal es mejorar la función del gen SMN2. Los bebés con diagnóstico de atrofia muscular espinal tienen al menos una copia del gen SMN2. Esta área ha sido investigada activamente por científicos estadounidenses, y actualmente se están realizando ensayos clínicos con varios medicamentos destinados a mejorar la síntesis de una proteína completa a partir del gen SMN2.
Otra vía de posible intervención terapéutica es explorar la neuroprotección para reducir la muerte de las neuronas motoras, aumentar su capacidad adaptativa y mejorar la funcionalidad.
La tercera dirección consiste en proteger el músculo de los procesos atróficos. Dado que la deficiencia de la proteína SMN afecta negativamente a las células nerviosas motoras y a la función muscular, el objetivo de este tratamiento debe ser proteger los músculos de la atrofia, aumentar la masa muscular y restaurar la función muscular. Este tipo de terapia no afectará el aparato genético, pero puede ralentizar o incluso bloquear el empeoramiento de la atrofia muscular espinal.
Detección de la atrofia muscular espinal
El cribado neonatal se utiliza cada vez más en la práctica médica y suele ser decisivo. Detectar la atrofia muscular espinal lo antes posible puede mejorar significativamente el pronóstico del niño enfermo. El cribado diagnóstico incluye los siguientes puntos, que se describen en la tabla:
Una forma de atrofia muscular espinal |
Sintomatología |
Atrofia muscular espinal tipo I (el niño no puede sentarse, esperanza de vida media: hasta 2 años) |
Se manifiesta desde el nacimiento hasta los seis meses. Se detecta un tono muscular insuficiente, el llanto es débil y la debilidad muscular (incluyendo la masticación y la deglución) aumenta. Hay problemas para retener la cabeza; el bebé adopta una postura de "rana" al acostarse. |
Atrofia muscular espinal tipo II (el niño puede sentarse, la esperanza de vida suele ser de más de 2 años y más de la mitad de los pacientes viven hasta los 20-25 años) |
Se presenta a partir de los 7 meses de edad y hasta el año y medio. En ocasiones, se observan problemas de deglución, respiratorios y tos. Los signos permanentes incluyen espasmos musculares, movilidad articular limitada, curvatura de la columna vertebral, presión arterial baja y debilidad muscular. |
Atrofia muscular espinal tipo III (el niño puede sentarse y moverse, pero las capacidades mencionadas se pierden gradualmente, la esperanza de vida es normal) |
Debuta al año y medio. Se observa curvatura de la columna vertebral y el tórax, atrofia muscular de la pelvis y la parte proximal de las piernas, y aumento de la movilidad articular. Presenta dificultad para tragar. |
Atrofia muscular espinal tipo IV |
Se refiere a la forma adulta. La sintomatología es muy similar a la de la atrofia muscular espinal tipo III. La debilidad aumenta gradualmente, y los temblores y las fascioculaciones musculares aparecen con la aparición entre los 16 y los 25 años. |
Pronóstico
En el síndrome de Werdnig-Hoffman, la esperanza de vida promedio es de 1,5 a 2 años. En la mayoría de los casos, el desenlace fatal se debe al aumento de la insuficiencia respiratoria y al desarrollo de inflamación pulmonar. Con apoyo respiratorio oportuno mediante ventilación artificial, es posible aumentar ligeramente la esperanza de vida del bebé. Existe una necesidad especial de cuidados paliativos continuos, que también se requieren en la amiotrofia espinal tipo II. Las patologías del tercer y cuarto tipo se caracterizan por un pronóstico más favorable.
Cualquier tipo de atrofia muscular espinal es una enfermedad grave. Todos los familiares del paciente requieren apoyo psicológico, informativo y social constante. Es importante que el paciente reciba un diagnóstico adecuado y apoyo profesional de especialistas como pediatras, neurólogos, neumólogos, cardiólogos, traumatólogos, fisioterapeutas, etc. A pesar de la falta de terapia específica para la enfermedad, se realiza tratamiento sintomático, se prescribe nutrición especial (tanto parenteral como enteral) y diversas medidas de rehabilitación que contribuyen a ralentizar la progresión de la patología y prevenir la aparición de complicaciones.
A muchos pacientes se les reconoce una discapacidad y se elabora un plan de rehabilitación individual.
La atrofia muscular espinal que se produce de forma natural sin el uso de equipos especiales para ayudar a la respiración y la alimentación termina en aproximadamente la mitad de los casos con la muerte del niño enfermo antes de los dos años de edad (principalmente enfermedad de tipo I).