Médico experto del artículo.

Nuevos artículos
Síndrome de Pierre Robin
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.

El síndrome de Pierre Robin, también conocido en medicina como anomalía de Robin, es una patología congénita del desarrollo de la mandíbula. La enfermedad recibe su nombre en honor al dentista francés P. Robin, quien describió por primera vez todos sus signos. Lannelongue y Menard describieron por primera vez el síndrome de Pierre Robin en 1891 en su informe sobre dos pacientes con micrognatia, paladar hendido y retroglosoptosis. En 1926, Pierre-Robin publicó un caso de la enfermedad en un lactante con signos del síndrome clásico. Hasta 1974, la tríada de signos se conocía como síndrome de Robin-Pierre. Sin embargo, este síndrome se utiliza actualmente para describir malformaciones con la presencia simultánea de múltiples anomalías.
Epidemiología
Se trata de un defecto congénito heterogéneo con una prevalencia de 1 por cada 8.500 nacidos vivos. La proporción hombre-mujer es de 1:1, excepto en la forma ligada al cromosoma X.
Entre estos pacientes, el 50% de los bebés tienen un paladar blando hendido incompleto, el resto nace con un paladar arqueado e inusualmente alto, pero sin hendidura.
Causas Síndrome de Pierre Robin
Se considera la posibilidad de herencia autosómica recesiva de la enfermedad. Existen dos tipos de síndrome según la etiología: aislado y genéticamente determinado. El tipo aislado se desarrolla debido a la compresión de la parte inferior de la mandíbula durante el desarrollo embrionario. La compresión puede desarrollarse debido a:
- Presencia de obstrucciones localizadas en el útero (quistes, cicatrices, tumores).
- Embarazo múltiple.
Además, el desarrollo de la mandíbula en el feto puede verse alterado por:
- Infecciones virales que sufrió la futura mamá durante el embarazo.
- Trastornos neurotróficos.
- Cantidad insuficiente de ácido fólico en el organismo de una mujer embarazada.
Patogenesia
El síndrome de Pierre Robin es causado por trastornos embrionarios que son provocados por una amplia variedad de patologías en el período prenatal.
Existen tres teorías fisiopatológicas que pueden explicar la aparición del síndrome de Pierre Robin.
Teoría mecánica: Esta teoría es la más probable. El subdesarrollo del aparato mandibular ocurre entre la 7.ª y la 11.ª semana de embarazo. La posición elevada de la lengua en la cavidad oral provoca la formación de hendiduras en el paladar, lo que impide el cierre de la vena cava. Esta teoría explica la clásica hendidura en forma de U invertida y la ausencia del labio hendido asociado. El oligohidramnios podría influir en la etiología, ya que la ausencia de líquido amniótico puede provocar la deformación del mentón y la consiguiente compresión de la lengua entre la vena cava.
Teoría neurológica: Se ha observado un retraso en el desarrollo neurológico con electromiografía de los músculos de la úvula y las columnas faríngeas, y del gusto debido al retraso de la conducción en el nervio hipogloso.
Teoría de la disneurorregulación del rombencéfalo: Esta teoría se basa en la alteración del desarrollo del rombencéfalo durante la ontogénesis.
El desarrollo insuficiente de la parte inferior de la mandíbula del niño provoca una reducción significativa de la cavidad oral. Esto, a su vez, causa la llamada pseudomacroglosia, es decir, el desplazamiento de la lengua hacia la parte posterior de la pared faríngea. Esta patología provoca la obstrucción de las vías respiratorias.
Mientras el bebé llora o se mueve, la vía aérea permanece despejada, pero tan pronto como el bebé se duerme, la obstrucción se produce nuevamente.
Debido a los trastornos respiratorios, la alimentación del bebé se dificulta mucho. En este momento, casi siempre se produce obstrucción de las vías respiratorias. Si no se aplica tratamiento médico, esta patología puede provocar un agotamiento grave de todo el cuerpo e incluso la muerte.
Síntomas Síndrome de Pierre Robin
La enfermedad se caracteriza por tres síntomas principales:
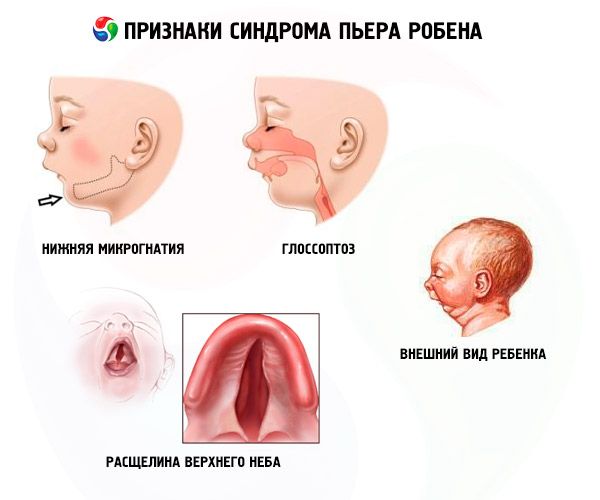
- La micrognatia inferior (subdesarrollo del maxilar inferior) se presenta en el 91,7 % de los casos. Se caracteriza por una retracción de la arcada dentaria inferior de 10 a 12 mm por detrás de la superior. El maxilar inferior presenta un cuerpo pequeño y un ángulo obtuso. El niño alcanza un desarrollo normal aproximadamente a los 5-6 años.
- Glosoptosis (retracción de la lengua debido a su desarrollo insuficiente, observada en el 70-85% de los casos).
- La macroglosia y la anquiloglosia son síntomas relativamente raros, observados en el 10-15% de los casos.
- Una grieta aparece en el cielo.
- Bradipnea y disnea.
- Cianosis leve.
- Asfixia, que ocurre con mayor frecuencia durante los intentos de alimentar al bebé.
- La tragación es imposible o muy difícil.
- Sensación de vomitar.
- Anomalías auriculares en el 75% de los casos.
- La pérdida auditiva conductiva se presenta en el 60% de los pacientes, mientras que la atresia del conducto auditivo externo se presenta sólo en el 5% de los pacientes, neumatización insuficiente de la cavidad mastoidea del hueso temporal.
- Anomalías del oído interno (aplasia de los canales semicirculares laterales, gran acueducto vestibular, pérdida de células ciliadas cocleares).
- Las malformaciones nasales son poco frecuentes y consisten principalmente en anomalías de la raíz nasal.
- Se presentan malformaciones dentales en el 30% de los casos. La laringomalacia y la insuficiencia velofaríngea se presentan en aproximadamente el 10-15% de los pacientes con síndrome de Pierre Robin.
Características sistémicas del síndrome de Pierre Robin
Las anomalías sistémicas del desarrollo se describen en el 10-85% de los casos registrados.
Las anomalías oculares se presentan en un 10-30% de los pacientes. Estas pueden incluir: hipermetropía, miopía, astigmatismo, esclerosis corneal y estenosis del conducto nasolagrimal.
Patologías cardiovasculares: soplos cardíacos benignos, estenosis de la arteria pulmonar, conducto arterioso persistente, ventana oval, comunicación interauricular e hipertensión pulmonar. Su prevalencia varía entre el 5% y el 58%.
Anomalías relacionadas con el sistema musculoesquelético (70-80% de los casos): sindactilia, falanges displásicas, polidactilia, clinodactilia, hipermovilidad articular y oligodactilia de las extremidades superiores. Anomalías de las extremidades inferiores: anomalías del pie (pie zambo, aducción metatarsiana), malformaciones femorales (pelvis en valgo o varo, fémures cortos), anomalías de la cadera (luxación congénita, contracturas), anomalías de la articulación de la rodilla (genu valgo, sincondrosis). Malformaciones de la columna vertebral: escoliosis, cifosis, lordosis, displasia vertebral, agenesia del sacro y del seno coccígeo.
Patología del sistema nervioso central: epilepsia, retrasos en el desarrollo del sistema nervioso, hidrocefalia. La frecuencia de defectos del SNC es de aproximadamente el 50%.
Anomalías genitourinarias: testículos no descendidos (25%), hidronefrosis (15%) e hidrocele (10%).
Síndromes y afecciones asociados: síndrome de Stickler, síndrome de trisomía 11q, trisomía 18, síndrome de deleción 4q, artropatía reumatoide, hipocondroplasia, síndrome de Moebius.
Etapa
Hay tres etapas de gravedad de la enfermedad, que dependen del estado del tracto respiratorio del niño:
- Leve: presenta leves problemas de alimentación, pero la respiración es prácticamente inexistente. El tratamiento es ambulatorio.
- Moderado: la respiración es moderadamente difícil, la alimentación del niño es moderadamente difícil. El tratamiento se realiza en un hospital.
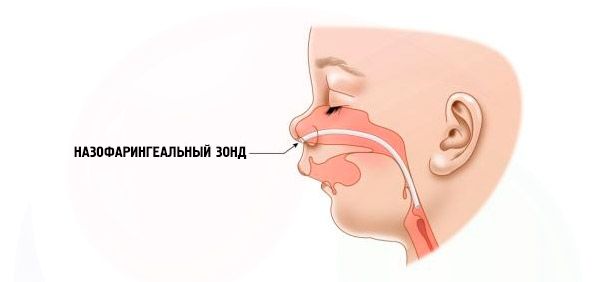
- Grave: la respiración es muy difícil y el niño no puede alimentarse con normalidad. Es necesario utilizar dispositivos especiales (sonda intranasal).
Complicaciones y consecuencias
La combinación de micrognatia y glosoptosis puede provocar complicaciones respiratorias graves y problemas durante la alimentación del niño.
El síndrome de Pierre Robin provoca las siguientes complicaciones:
- Respiración estridótica por obstrucción de las vías respiratorias. Laringomalacia o incluso asfixia durante el sueño.
- El desarrollo psicomotor del niño está muy por detrás del de sus compañeros.
- El desarrollo físico también está rezagado.
- El habla de los pacientes está alterada.
- Infecciones de oído frecuentes que se vuelven crónicas y provocan pérdida auditiva.
- Síndrome de apnea obstructiva del sueño, la aparición de muerte durante el sueño varía en el 14-91% de los casos.
- Problemas con los dientes.
Diagnostico Síndrome de Pierre Robin
El diagnóstico del síndrome de Pierre Robin no es difícil. Se basa en las manifestaciones clínicas. Para descartar otras patologías, es fundamental consultar con un genetista.
Los niños con anomalía congénita de Robin presentan problemas respiratorios desde el nacimiento debido a la constante caída de la lengua. El bebé está inquieto, su piel está azulada y presenta sibilancias al inhalar. Puede atragantarse durante la alimentación. El diagnóstico también se puede realizar por la apariencia inusual del niño: "cara de pájaro". Con frecuencia, los pacientes desarrollan otros defectos: miopía, cataratas, patología del sistema genitourinario, cardiopatía y anomalías en el desarrollo de la columna vertebral.
En base a estas manifestaciones clínicas no será difícil para un especialista realizar un diagnóstico correcto.
¿A quién contactar?
Tratamiento Síndrome de Pierre Robin
El tratamiento se realiza inmediatamente después del nacimiento de un niño con síndrome de Pierre Robin. Si la enfermedad es leve, para mejorar el estado del paciente es necesario mantener al niño en posición vertical o boca abajo constantemente. La cabeza del bebé debe estar inclinada hacia el pecho. Durante la alimentación, no se recomienda mantener al niño en posición horizontal para evitar que los alimentos entren en las vías respiratorias.
Si el subdesarrollo de la mandíbula inferior es muy pronunciado, se recurre a la intervención quirúrgica para restituir la lengua retraída a su posición fisiológica normal. En casos graves, se levanta la lengua y se fija al labio inferior. En casos muy graves, se debe realizar una traqueotomía, glosopexia y osteogénesis por distracción de la mandíbula inferior.
También se utiliza el tratamiento conservador.
Medicamentos
Fenobarbital. Es un somnífero y sedante con efecto anticonvulsivo. Cada comprimido contiene 100 ml de fenobarbital. La dosis es individual, ya que depende de la gravedad de la enfermedad y del estado del niño. Este medicamento está contraindicado en pacientes con insuficiencia hepática, hipercinesia, anemia, miastenia, porfiria, diabetes mellitus, depresión e intolerancia a los componentes. Al tomarlo, pueden presentarse los siguientes síntomas: mareos, astenia, alucinaciones, agranulocitosis, náuseas, hipotensión y alergias.
Clonazepam. Medicamento recetado para el tratamiento de la epilepsia. Contiene el principio activo clonazepam, un derivado de las benzodiazepinas. Tiene efectos anticonvulsivos, ansiolíticos y relajantes musculares. La dosis la determina el médico tratante, pero no debe exceder el máximo de 250 mcg al día. No lo tome en caso de insomnio, hipertonía muscular, agitación psicomotora o trastornos de pánico. Al tomarlo, pueden presentarse los siguientes síntomas: letargo, náuseas, dismenorrea, cefalea, leucopenia, retención o incontinencia urinaria, alopecia y alergia.
Sibazón. Disponible en solución y comprimidos rectales. El principio activo es un derivado de la benzodiazepina (sibazón). Tiene efecto sedante, ansiolítico y anticonvulsivo. La dosis es individual. Se prohíbe el uso de este medicamento en pacientes con hipercapnia crónica, miastenia grave o intolerancia a las benzodiazepinas. Al tomarlo, pueden presentarse los siguientes síntomas: náuseas, estreñimiento, dolor de cabeza, mareos, hipo, incontinencia urinaria y alergias.
Liofilizado de Cortexin. Medicamento con efecto nootrópico. Contiene un complejo de fracciones polipeptídicas hidrosolubles y glicina. La dosis es individual y la prescribe el médico tratante según la condición del paciente. Se prohíbe el uso de Cortexin a pacientes con intolerancia. Puede causar reacciones alérgicas.
Tratamiento de fisioterapia
Por lo general, en las etapas leves del síndrome se utiliza la terapia posicional, donde se coloca al niño boca abajo en posición vertical hasta que la gravedad fuerza a la mandíbula inferior a crecer correctamente.
Tratamiento quirúrgico
El tratamiento quirúrgico se utiliza principalmente para corregir la glosoptosis. Existen varios métodos:
- Se sujeta la lengua con un hilo de plata. El hilo se pasa por la parte inferior de la encía y el labio inferior. El método se llama Douglas.
- Método de Duhamel: se pasa un hilo grueso de plata por la base de la lengua y ambas mejillas del paciente. No se debe usar durante más de treinta días.
- Dispositivos ortopédicos para extensión y fijación de la lengua.
- Al año de edad se puede realizar una cirugía para corregir el paladar hendido.
Pronóstico
El pronóstico y la evolución de la enfermedad son graves. Con mayor frecuencia, la muerte ocurre en los primeros días de vida, en la etapa moderada o grave de la enfermedad (la causa es asfixia). Además, el riesgo de muerte durante el primer año es bastante alto debido a numerosas infecciones.
Para los pacientes mayores de dos años, el pronóstico es favorable.
 [ 36 ]
[ 36 ]