Médico experto del artículo.

Nuevos artículos
Polimiositis y dermatomiositis: causas, síntomas, diagnóstico, tratamiento
Último revisado: 05.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
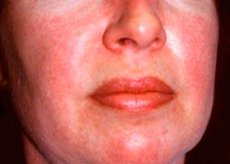
La polimiositis y la dermatomiositis son enfermedades reumáticas sistémicas poco frecuentes que se caracterizan por cambios inflamatorios y degenerativos en los músculos (polimiositis) o en los músculos y la piel (dermatomiositis). La manifestación cutánea más específica es la erupción en heliotropo.
La afectación muscular es simétrica e incluye debilidad, algo de dolor a la palpación y atrofia subsiguiente de los músculos proximales de la cintura pélvica. Las complicaciones pueden incluir afectación visceral y neoplasia maligna. El diagnóstico se basa en la presentación clínica y la evaluación de la disfunción muscular mediante la medición de los niveles enzimáticos, la realización de resonancia magnética, electromiografía y biopsia muscular. El tratamiento consiste en glucocorticoides, a veces en combinación con inmunosupresores o inmunoglobulinas intravenosas.
Las mujeres se enferman con el doble de frecuencia que los hombres. La enfermedad puede presentarse a cualquier edad, pero se detecta con mayor frecuencia entre los 40 y los 60 años; en niños, entre los 5 y los 15 años.
 [
[ ¿Qué causa la dermatomiositis y la polimiositis?
Se cree que la causa de la enfermedad es una reacción autoinmune al tejido muscular en individuos genéticamente predispuestos. La enfermedad es más común en presencia de antecedentes familiares con alta carga de enfermedad y portadores de ciertos antígenos HLA (DR3, DR52, DR56). Los posibles desencadenantes son la miositis viral y las neoplasias malignas. Existen informes de la detección de estructuras similares a los picornavirus en células musculares; además, los virus pueden inducir enfermedades similares en animales. La asociación de tumores malignos con la dermatomiositis (mucho menos frecuente que con la polimiositis) sugiere que el crecimiento tumoral también puede ser un desencadenante del desarrollo de la enfermedad como resultado del inicio de reacciones autoinmunes a antígenos comunes del tumor y del tejido muscular.
Se encuentran depósitos de IgM, IgG y el tercer componente del complemento en las paredes de los vasos sanguíneos del músculo esquelético; esto es especialmente característico de la dermatomiositis en niños. Los pacientes con polimiositis también pueden desarrollar otros procesos autoinmunes.
Fisiopatología de la dermatomiositis y la polimiositis
Los cambios patológicos incluyen daño celular y atrofia en el contexto de inflamación de diversa gravedad. Los músculos de las extremidades superiores e inferiores, así como los de la cara, se dañan en menor medida que otros músculos esqueléticos. El daño a los músculos viscerales de la faringe y el esófago superior, y con menor frecuencia al corazón, el estómago o los intestinos, puede provocar disfunción de estos órganos. Las altas concentraciones de mioglobina debidas a la rabdomiólisis pueden causar daño renal. También pueden presentarse cambios inflamatorios en las articulaciones y los pulmones, especialmente en pacientes con anticuerpos antisintetasa.
Síntomas de la dermatomiositis y la polimiositis
La polimiositis puede aparecer de forma aguda (especialmente en niños) o subaguda (con mayor frecuencia en adultos). Una infección viral aguda a veces precede o desencadena la manifestación de la enfermedad, cuyas manifestaciones más comunes son la debilidad de los músculos proximales o las erupciones cutáneas. El dolor se manifiesta en menor medida que la debilidad. Pueden presentarse poliartralgia, fenómeno de Raynaud, disfagia, trastornos pulmonares y síntomas generales (aumento de la temperatura corporal, disminución del peso corporal, debilidad). El fenómeno de Raynaud se observa con frecuencia en pacientes con enfermedades concomitantes del tejido conectivo.
La debilidad muscular puede progresar durante varias semanas o meses. Sin embargo, para que se manifieste clínicamente, debe estar afectada al menos el 50 % de las fibras musculares (por lo tanto, la presencia de debilidad muscular indica la progresión de la miositis). Los pacientes pueden experimentar dificultad para levantar los brazos por encima del nivel de los hombros, subir escaleras o levantarse de una posición sentada. Debido a la grave debilidad de los músculos pélvicos y de la cintura escapular, los pacientes pueden verse confinados a una silla de ruedas o a una cama. Si se ven afectados los flexores del cuello, resulta imposible levantar la cabeza de la almohada. La afectación de los músculos de la faringe y el esófago superior provoca trastornos de la deglución y regurgitación. Los músculos de las extremidades inferiores y superiores, así como de la cara, no suelen verse afectados. Sin embargo, pueden desarrollarse contracturas en las extremidades.
Las erupciones cutáneas observadas en la dermatomiositis suelen ser de color oscuro y eritematosas. El edema periorbitario de color púrpura (erupción en heliotropo) también es característico. Las erupciones cutáneas pueden estar ligeramente elevadas por encima del nivel de la piel y ser lisas o escamosas; la localización de las erupciones es la frente, el cuello, los hombros, el pecho, la espalda, los antebrazos, las partes inferiores de las espinillas, las cejas, las áreas de las rodillas, los maléolos mediales, las superficies dorsales de las articulaciones interfalángicas y metacarpofalángicas, en el lado lateral (síntoma de Gottron). Es posible la hiperemia de la base o la periferia de las uñas. La dermatitis descamativa, acompañada de grietas, puede desarrollarse en la piel de la superficie lateral de los dedos. Las lesiones cutáneas primarias a menudo se resuelven sin secuelas, pero pueden conducir a cambios secundarios como pigmentación oscura, atrofia, cicatrización o vitíligo. Pueden desarrollarse calcificaciones subcutáneas, especialmente en niños.
Aproximadamente el 30% de los pacientes desarrollan poliartralgia o poliartritis, a menudo acompañada de edema y derrame articular. Sin embargo, la gravedad de las manifestaciones articulares es baja. Se presentan con mayor frecuencia cuando se detectan anticuerpos contra Jo-1 u otras sintetasas.
La afectación de órganos internos (excepto la faringe y el esófago superior) es menos frecuente en la polimiositis que en otras enfermedades reumáticas (en particular, el LES y la esclerosis sistémica). En raras ocasiones, sobre todo en el síndrome antisintetasa, la enfermedad se manifiesta como neumonitis intersticial (con disnea y tos). Pueden presentarse arritmias cardíacas y trastornos de la conducción, pero suelen ser asintomáticos. Las manifestaciones gastrointestinales son más frecuentes en niños con vasculitis y pueden incluir vómitos con sangre, melena y perforación intestinal.
¿Donde duele?
Clasificación de la polimiositis
Hay 5 tipos de polimiositis.
- La polimiositis idiopática primaria, que puede ocurrir a cualquier edad, no afecta la piel.
- La dermatomiositis idiopática primaria es similar a la polimiositis idiopática primaria pero afecta la piel.
- La polimiositis y la dermatomiositis asociadas con neoplasias malignas pueden presentarse en pacientes de cualquier edad; su desarrollo se observa con mayor frecuencia en pacientes de edad avanzada, así como en pacientes con otras enfermedades del tejido conectivo. El desarrollo de neoplasias malignas puede observarse tanto en los 2 años previos como en los 2 años posteriores a la aparición de la miositis.
- La polimiositis o dermatomiositis infantil se asocia con vasculitis sistémica.
- La polimiositis y la dermatomiositis también pueden presentarse en pacientes con otras enfermedades del tejido conectivo, las más comunes son la esclerosis sistémica progresiva, la enfermedad mixta del tejido conectivo y el LES.
La inclusión de la miositis de los músculos del tronco en el grupo de la polimiositis es incorrecta, ya que esta última es una enfermedad independiente que se caracteriza por manifestaciones clínicas similares a las de la polimiositis idiopática crónica. Sin embargo, se desarrolla en la vejez, suele afectar los músculos de las partes distales del cuerpo (por ejemplo, las extremidades superiores e inferiores), tiene una mayor duración, responde peor al tratamiento y se caracteriza por un cuadro histológico típico.
Diagnóstico de la dermatomiositis y la polimiositis
Se debe sospechar polimiositis en pacientes con debilidad muscular proximal, con o sin dolor a la palpación. Es necesario evaluar la dermatomiositis en pacientes con exantema similar al heliotropo o al signo de Gottron, así como en pacientes con manifestaciones de polimiositis combinadas con lesiones cutáneas compatibles con dermatomiositis. Las manifestaciones clínicas de la polimiositis y la dermatomiositis pueden ser similares a las de la esclerosis sistémica o, con menor frecuencia, al LES o la vasculitis. La certeza del diagnóstico aumenta si se cumplen tantos de los siguientes cinco criterios como sea posible:
- debilidad de los músculos proximales;
- erupciones cutáneas características;
- aumento de la actividad de las enzimas del tejido muscular (creatina quinasa o, en ausencia de un aumento de su actividad, aminotransferasas o aldolasa);
- cambios característicos en la miografía o resonancia magnética;
- cambios histológicos característicos en una biopsia de tejido muscular (criterio absoluto).
La biopsia muscular permite descartar algunas afecciones clínicamente similares, como la miositis de los músculos del tronco y la rabdomiólisis por infección viral. Los cambios observados en el examen histológico pueden variar, pero la inflamación crónica y los focos de degeneración y regeneración muscular son típicos. Es necesario un diagnóstico preciso (generalmente mediante verificación histológica) antes de iniciar un tratamiento potencialmente tóxico. La resonancia magnética puede detectar focos de edema e inflamación en los músculos, seguida de su biopsia dirigida.
Los estudios de laboratorio pueden confirmar o, por el contrario, descartar la sospecha de la enfermedad, y también son útiles para evaluar su gravedad, la posibilidad de combinación con otras patologías similares y el diagnóstico de complicaciones. A pesar de que se detectan anticuerpos antinucleares en algunos pacientes, este fenómeno es más típico de otras enfermedades del tejido conectivo. Alrededor del 60% de los pacientes presentan anticuerpos contra el antígeno de núcleos (PM-1) o células completas del timo y Jo-1. El papel de los autoanticuerpos en la patogénesis de la enfermedad sigue sin estar claro, aunque se sabe que los anticuerpos contra Jo-1 son un marcador específico del síndrome antisintetasa, que incluye alveolitis fibrosante, fibrosis pulmonar, artritis y fenómeno de Raynaud.
La evaluación periódica de la actividad de la creatinquinasa es útil para monitorizar el tratamiento. Sin embargo, en pacientes con atrofia muscular grave, la actividad enzimática puede ser normal a pesar de la presencia de miositis crónica activa. La resonancia magnética, la biopsia muscular o una actividad elevada de la creatinquinasa suelen ser útiles para diferenciar entre una recaída de polimiositis y una miopatía inducida por glucocorticoides.
Dado que muchos pacientes presentan neoplasias malignas no diagnosticadas, algunos autores recomiendan realizar pruebas de detección a todos los adultos con dermatomiositis y polimiositis mayores de 60 años mediante el siguiente esquema: exploración física, que incluye tactos mamarios, pélvicos y rectales (incluida la prueba de sangre oculta en heces); hemograma completo; bioquímica sanguínea; mamografía; determinación del antígeno carcinoembrionario; análisis de orina; radiografía de tórax. Algunos autores cuestionan la necesidad de realizar estas pruebas de detección en pacientes más jóvenes sin evidencia clínica de neoplasia maligna.
¿Qué es necesario examinar?
Tratamiento de la dermatomiositis y la polimiositis
Hasta que se alivie la inflamación, se debe limitar la actividad física. Los glucocorticoides son fármacos de primera línea. En la fase aguda de la enfermedad, a los pacientes adultos se les debe recetar prednisolona (por vía oral) en dosis de 40 a 60 mg al día. La determinación regular de la actividad de la creatinquinasa es un indicador temprano de eficacia: en la mayoría de los pacientes, se observa una disminución o normalización entre 6 y 12 semanas después de un aumento de la fuerza muscular. Tras la normalización de la actividad enzimática, se reduce la dosis de prednisolona: primero en aproximadamente 2,5 mg al día durante una semana, y luego más rápidamente; si el aumento de la actividad enzimática muscular recae, se aumenta de nuevo la dosis hormonal. Los pacientes recuperados pueden prescindir de los glucocorticoides, pero con mayor frecuencia los pacientes adultos requieren terapia con glucocorticoides a largo plazo (10-15 mg de prednisolona al día). La dosis inicial de prednisolona para niños es de 30 a 60 mg/m² una vez al día. En presencia de remisión durante > 1 año en niños, se puede suspender el tratamiento con glucocorticoides.
En algunos casos, los pacientes que reciben altas dosis de glucocorticoides experimentan un aumento repentino de la debilidad muscular, que puede estar asociada con el desarrollo de miopatía por glucocorticoides.
En caso de respuesta inadecuada al tratamiento con glucocorticoides, así como de desarrollo de miopatía por glucocorticoides u otras complicaciones que requieran reducción de la dosis o la suspensión de la prednisolona, se deben utilizar inmunosupresores (metotrexato, ciclofosfamida, azatioprina, ciclosporina). Algunos pacientes pueden recibir solo metotrexato (generalmente en dosis superiores a las utilizadas para el tratamiento de la AR) durante más de 5 años. Las inmunoglobulinas intravenosas pueden ser eficaces en pacientes refractarios al tratamiento farmacológico, pero su uso incrementa el coste del tratamiento.
La miositis asociada a tumores primarios y metastásicos, así como la miositis de los músculos del tronco, suelen ser más refractarias al tratamiento con glucocorticoides. La remisión de la miositis asociada a tumores malignos es posible tras la extirpación del tumor.
¿Cuál es el pronóstico de la dermatomiositis y la polimiositis?
Se observa una remisión a largo plazo (e incluso recuperación clínica) de más de 5 años en más de la mitad de los pacientes tratados; en niños, esta cifra es mayor. Sin embargo, la recaída puede ocurrir en cualquier momento. La tasa de supervivencia general a cinco años es del 75 %, mayor en niños. Las causas de muerte en adultos son debilidad muscular grave y progresiva, disfagia, desnutrición, neumonía por aspiración o insuficiencia respiratoria por infecciones pulmonares. La polimiositis es más grave y resistente al tratamiento si hay daño cardíaco y pulmonar. La muerte en niños puede ocurrir debido a vasculitis intestinal. El pronóstico general de la enfermedad también depende de la presencia de neoplasias malignas.