Médico experto del artículo.

Nuevos artículos
Síndrome de Aper
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.

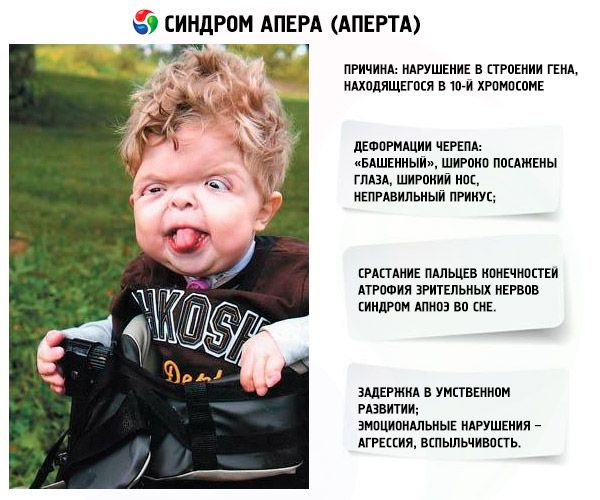
El síndrome de Apert es una patología genética que produce defectos en el desarrollo del cráneo (ojos muy separados, cráneo excesivamente alto) y, además, de las extremidades inferiores y superiores (los dedos de las mismas están completamente fusionados; también pueden aparecer dedos adicionales).
 [
[ Causas Síndrome de Aper
El desarrollo del síndrome es provocado por un trastorno en la estructura de un gen localizado en el cromosoma 10. Este gen es responsable del proceso de separación de los dedos, y además, del cierre oportuno de las suturas craneales.
Además, las enfermedades infecciosas sufridas durante el embarazo (como rubéola, tuberculosis, sífilis, gripe y meningitis) y la exposición de la madre a rayos X se consideran causas de la patología. Con mayor frecuencia, este síndrome se detecta en hijos de padres mayores.
Patogenesia
El síndrome de Apert es hereditario. Su tipo es autosómico dominante (es decir, en una familia donde uno de los futuros padres está enfermo, la probabilidad de tener un bebé con esta enfermedad es del 50 al 100%).
Una mutación única en el receptor 2 del factor de crecimiento de fibroblastos (FGFR2) produce un mayor número de células progenitoras que se desarrollan a lo largo de la vía osteogénica. Esto, en última instancia, resulta en una mayor formación de matriz ósea subperióstica y una osificación prematura de las suturas craneales durante el desarrollo fetal. El orden y la velocidad de fusión de las suturas determinan el grado de deformidad y discapacidad. Una vez cicatrizada la sutura, se limita el crecimiento de otros tejidos perpendiculares a ella, y los huesos fusionados actúan como un único andamiaje óseo.
La primera evidencia genética de que la sindactilia en el síndrome de Apert se debe a un defecto en el receptor del factor de crecimiento de queratinocitos (KGFR) fue la observación de una correlación entre la expresión de KGFR en fibroblastos y la gravedad de la sindactilia.
La ambliopía y el estrabismo son más frecuentes en pacientes con la mutación Ser252Trp del FGFR2, y la atrofia del disco óptico es más frecuente en pacientes con la mutación Pro253Arg del FGFR2. Los pacientes con mutaciones Ser252Trp del FGFR2 presentan una prevalencia significativamente mayor de discapacidad visual en comparación con los pacientes con la mutación Pro253Arg del FGFR2.
Síntomas Síndrome de Aper
Algunas manifestaciones de la enfermedad son claramente perceptibles ya en el nacimiento del bebé, ya que se desarrollan dentro del útero materno. Entre los principales síntomas del síndrome se encuentran:
- El cráneo se deforma, se estira en altura y adquiere una apariencia de "torre"; además, los ojos están muy separados y ligeramente abultados (debido a que el tamaño de las cuencas oculares disminuye); la nariz se ensancha y se forma una mordida incorrecta (los dientes superiores sobresalen excesivamente);
- Los dedos de las extremidades están completamente fusionados (principalmente el dedo anular, así como el dedo medio y el índice) y parecen una membrana de piel o una fusión completa de huesos; además, pueden crecer dedos adicionales;
- Retraso mental (no se presenta en todos los casos);
- Los nervios ópticos se atrofian, como resultado de lo cual disminuye la agudeza visual (en algunos casos, se desarrolla una pérdida completa de la visión);
- El aumento de la presión intracraneal, que se produce como resultado del cierre prematuro de las suturas craneales, se manifiesta en forma de dolores de cabeza y vómitos con náuseas;
- Como la mandíbula superior permanece subdesarrollada, surgen problemas respiratorios;
- El síndrome de apnea del sueño es una ocurrencia común.
- Manifestaciones emocionales: agresión, falta de control, fuerte irascibilidad.
Complicaciones y consecuencias
La mayoría de los pacientes presentan algún grado de obstrucción de las vías respiratorias superiores durante la infancia. La vía aérea superior presenta una permeabilidad deficiente debido al tamaño reducido de la nasofaringe y las coanas, y la vía aérea inferior puede causar muerte prematura debido a anomalías del cartílago traqueal.
Diagnostico Síndrome de Aper
Para realizar un diagnóstico se requieren los siguientes pasos:
- El médico debe analizar las quejas del paciente, así como su historial médico. Es necesario determinar si ha habido casos de patología similar en la familia.
- Examen neurológico para evaluar la forma del cráneo y el desarrollo intelectual del paciente (cuestionarios especiales y entrevista);
- Examen del fondo de ojo para detectar la presencia de síntomas de aumento de la presión intracraneal (hinchazón del disco óptico, así como desenfoque de sus bordes);
- Para evaluar el estado del cráneo se realiza una radiografía;
- Imágenes computarizadas y resonancia magnética de la cabeza para examinar la estructura del cerebro y del cráneo capa por capa, para determinar la presencia de síntomas de fusión prematura de las suturas craneales y, además, hidrocefalia (debido al aumento de la presión intracraneal, se acumula un exceso de líquido cefalorraquídeo (este es el líquido cefalorraquídeo que promueve el proceso metabólico, así como la nutrición del cerebro));
- Radiografía de pies y manos para determinar la causa de la fusión de los dedos (esto es importante para planificar la intervención quirúrgica posterior);
- Se podría prescribir una consulta con un médico genetista y un neurocirujano.
Pruebas
Se está realizando un análisis genético de las mutaciones comunes que ocurren en el gen tipo FGFR2.
Diagnóstico diferencial
Este síndrome debe diferenciarse de otras patologías genéticas con craneosinostosis, como los síndromes de Pfeiffer, Crouzon, Saethre-Chotzen y Carpenter. Se utilizan métodos de análisis genético molecular para descartar estas anomalías.
¿A quién contactar?
Tratamiento Síndrome de Aper
La cirugía se considera el único tratamiento eficaz para el síndrome de Apert: ayuda a corregir defectos físicos individuales y también corrige el retraso mental.
Durante este procedimiento, se cierra la sutura coronal para prevenir posibles lesiones cerebrales. La técnica más común es la distracción craneofacial, que implica una tracción gradual del cráneo. Se realiza cirugía ortodóncica y/o ortognática para eliminar defectos faciales individuales.
Además, los pacientes se someten a una corrección quirúrgica de la fusión de los dedos.
Referencias
Genética Médica. Guía Nacional. Editado por Ginter EK, Puzyrev VP GEOTAR-Media. 2022