Médico experto del artículo.

Nuevos artículos
Síndrome de Usher
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
El síndrome de Usher es una enfermedad hereditaria que se manifiesta con sordera total de nacimiento y ceguera progresiva con la edad. La pérdida de visión se asocia con la retinosis pigmentaria, un proceso de degeneración pigmentaria de la retina. Muchas personas con síndrome de Usher también presentan graves problemas de equilibrio.
Epidemiología
Gracias a la investigación, se pudo establecer que el síndrome de Usher afecta a aproximadamente el 8% de los niños sordomudos examinados (las pruebas se realizaron en instituciones especializadas para sordomudos). Se observó retinitis pigmentaria en el 6-10% de los pacientes con sordera congénita, la cual, a su vez, se observa en aproximadamente el 30% de las personas con enfermedad pigmentaria de la retina.
Se cree que esta enfermedad se manifiesta en aproximadamente 3 a 10 personas de cada 100 mil en todo el mundo. Se observa por igual tanto en mujeres como en hombres. Entre el 5% y el 6% de la población mundial padece este síndrome. Cerca del 10% de los casos de sordera profunda infantil se deben al síndrome de Usher tipo I y tipo II.
En Estados Unidos, los tipos 1 y 2 son los más comunes. En conjunto, representan aproximadamente entre el 90 % y el 95 % de todos los casos de síndrome de Usher en niños.
Causas Síndrome de Usher
El síndrome de Usher tipos I, II y III tiene una causa autosómica recesiva, mientras que el tipo IV se considera un trastorno del cromosoma X. Las causas de ceguera y sordera asociadas a este síndrome aún no se han estudiado suficientemente. Se supone que las personas con esta enfermedad presentan hipersensibilidad a componentes que pueden dañar la estructura del ADN. Además, esta enfermedad puede estar asociada a trastornos del sistema inmunitario, pero en este caso, no existe una descripción precisa de este proceso.
En 1989, se identificaron por primera vez anomalías cromosómicas en pacientes con enfermedad de tipo II, lo que en el futuro podría permitir aislar los genes que causan el síndrome. También podría ser posible identificar estos genes en portadores y desarrollar pruebas genéticas prenatales especiales.
 [ 8 ]
[ 8 ]
Factores de riesgo
El síndrome se hereda cuando ambos progenitores están afectados, es decir, se hereda de forma recesiva. Un hijo también puede heredar la enfermedad si sus padres son portadores del gen. Si ambos futuros progenitores tienen este gen, la probabilidad de tener un bebé con este síndrome es de 1 en 4. Una persona que solo tiene un gen del síndrome se considera portadora, pero no presenta síntomas del trastorno. Actualmente, aún no es posible determinar si una persona tiene el gen de esta enfermedad.
Si un niño nace de padres uno de los cuales no tiene ese gen, entonces la probabilidad de que herede el síndrome es muy baja, pero definitivamente será portador.
Síntomas Síndrome de Usher
Los síntomas del síndrome de Usher incluyen pérdida auditiva y acumulación anormal de células pigmentadas en las estructuras oculares. Posteriormente, el paciente desarrolla degeneración de la retina, lo que provoca deterioro de la visión y, en los casos más graves, pérdida de la visión.
La pérdida auditiva neurosensorial puede ser leve o completa y, por lo general, no progresa desde el nacimiento. Sin embargo, la enfermedad pigmentaria de la retina puede comenzar a desarrollarse en la infancia o más tarde. Los resultados de las pruebas han demostrado que la agudeza visual central puede mantenerse durante muchos años, incluso cuando la visión periférica se deteriora (una afección denominada "visión de túnel").
Éstas son las principales manifestaciones de la enfermedad, que en ocasiones pueden complementarse con otros trastornos, como psicosis y otros trastornos mentales, problemas en el oído interno y/o cataratas.
Formas
Durante la investigación se identificaron tres tipos de esta enfermedad, así como una cuarta forma, que es bastante rara.
El tipo I de la enfermedad se caracteriza por sordera congénita completa y trastornos del equilibrio. A menudo, estos niños comienzan a caminar solo al año y medio de edad. El deterioro de la visión suele comenzar a los 10 años, y la ceguera nocturna se manifiesta finalmente a los 20 años. Los niños con este tipo de enfermedad pueden desarrollar un deterioro progresivo de la visión periférica.
En la enfermedad de tipo II, se observa sordera moderada o congénita. En este caso, el deterioro de la sordera parcial suele desaparecer. La retinitis pigmentaria comienza a desarrollarse hacia el final de la adolescencia o después de los 20 años. La ceguera nocturna suele comenzar entre los 29 y los 31 años. La pérdida de agudeza visual en la patología de tipo II suele progresar un poco más lentamente que en la de tipo I.
El tipo III de la enfermedad se caracteriza por una pérdida auditiva progresiva, que generalmente comienza durante la pubertad, así como por el desarrollo gradual durante el mismo período (un poco más tarde que la pérdida auditiva) de retinitis pigmentosa, que puede convertirse en un factor en el desarrollo de ceguera progresiva.
Las manifestaciones de la patología de tipo IV se presentan principalmente en varones. En este caso, también se observan trastornos progresivos y pérdida de audición y visión. Esta forma es muy poco frecuente y suele ser de origen cromosómico X.
Diagnostico Síndrome de Usher
El diagnóstico del síndrome de Usher se realiza basándose en la combinación observada en el paciente de sordera repentina y pérdida progresiva de la visión.
Pruebas
Es posible que se solicite una prueba genética especial para detectar la mutación.
Se han encontrado once loci genéticos que pueden provocar el desarrollo del síndrome de Usher y se han identificado nueve genes que son definitivamente la causa del trastorno:
- Tipo 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Tipo 2: ush2a, VLGR1, WHRN.
- Síndrome de Usher tipo 3: USH3A.
Los científicos del NIDCD, junto con colegas de universidades de Nueva York e Israel, han identificado una mutación llamada R245X en el gen Pcdh15 que representa un gran porcentaje del síndrome de Usher tipo 1 en la población judía.
Para obtener información sobre los laboratorios que realizan ensayos clínicos, visite https://www.genetests.org y busque en el directorio de laboratorios "síndrome de Usher".
Para obtener más información sobre los ensayos clínicos existentes que incluyen pruebas genéticas para el síndrome de Usher, visite https://www.clinicaltrials.gov y busque "síndrome de Usher" o "pruebas genéticas del síndrome de Usher".
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Diagnóstico instrumental
Existen varios métodos de diagnóstico instrumental:
- Examen del fondo de ojo para detectar la presencia de manchas pigmentarias en la retina, así como estrechamiento de los vasos retinianos;
- Electrorretinograma, que permite detectar desviaciones degenerativas iniciales en la retina. Muestra la extinción de las vías electrorradiográficas.
- Un electronistagmograma (ENG) mide los movimientos oculares involuntarios que podrían indicar la presencia de un desequilibrio.
- Audiometría, que se utiliza para determinar la presencia de sordera y su gravedad.
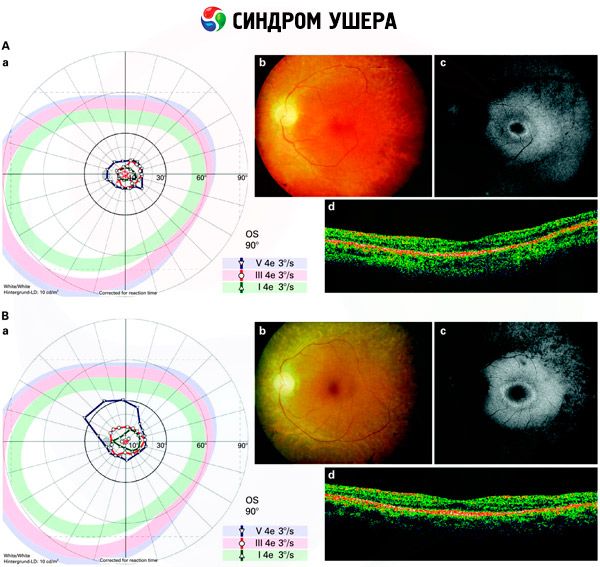
Diagnóstico diferencial
El síndrome de Usher debe diferenciarse de algunos trastornos similares.
Síndrome de Hallgren, que se caracteriza por pérdida auditiva congénita y pérdida progresiva de la visión (también se desarrollan cataratas y nistagmo). Otros síntomas incluyen ataxia, trastornos psicomotores, psicosis y retraso mental.
El síndrome de Alström es una enfermedad hereditaria que provoca la degeneración de la retina, lo que resulta en la pérdida de la visión central. Este síndrome se asocia con la obesidad infantil. Simultáneamente, la diabetes mellitus y la pérdida auditiva comienzan a desarrollarse después de los 10 años.
La rubéola en una mujer embarazada durante el primer trimestre puede causar diversas anomalías en el desarrollo del niño. Entre las consecuencias de esta anomalía se encuentran la pérdida de audición, así como problemas de visión y, además, diversos defectos del desarrollo.
¿A quién contactar?
Tratamiento Síndrome de Usher
Actualmente no existe cura para el síndrome de Usher. Por lo tanto, la terapia en este caso consiste principalmente en ralentizar el proceso de pérdida de visión y compensar la pérdida de audición. Los posibles métodos de tratamiento incluyen:
- Tomar vitamina A (algunos oftalmólogos creen que dosis altas de palmitato de vitamina A pueden retardar, pero no detener, la progresión de la retinitis pigmentosa);
- Implantación de dispositivos electrónicos especiales en los oídos del paciente (audífonos, implantes cocleares).
Los oftalmólogos recomiendan que la mayoría de los adultos con formas comunes de retinosis pigmentaria tomen 15,000 UI (unidades internacionales) de palmitato de vitamina A diariamente bajo supervisión. Dado que el estudio no incluyó a personas con síndrome de Usher tipo 1, no se recomiendan dosis altas de vitamina A para este grupo de pacientes. Quienes estén considerando tomar vitamina A deben consultar esta opción de tratamiento con su médico. Otras recomendaciones para esta opción de tratamiento incluyen:
- Cambiar su dieta para incluir alimentos ricos en vitamina A.
- Las mujeres que planean quedar embarazadas deben dejar de tomar dosis altas de vitamina A tres meses antes de concebir debido a un mayor riesgo de defectos congénitos.
- Las mujeres embarazadas deben dejar de tomar dosis altas de vitamina A debido al mayor riesgo de defectos congénitos.
También es importante adaptar al niño a la vida social. Esto requiere la ayuda de educadores especiales y psicólogos. Si el paciente ha comenzado a experimentar pérdida progresiva de la visión, se le debe enseñar a usar la lengua de señas.
Pronóstico
El síndrome de Usher tiene un pronóstico desfavorable. El campo visual y su agudeza comienzan a deteriorarse entre los 20 y los 30 años en la mayoría de los pacientes con esta enfermedad, independientemente de su tipo. En algunos casos, se produce pérdida total de la visión bilateral. La pérdida auditiva, que siempre se acompaña de mudez, evoluciona rápidamente hasta convertirse en pérdida auditiva bilateral completa.