Médico experto del artículo.

Nuevos artículos
Mucopolisacaridosis tipo 3
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
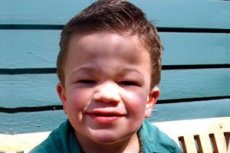
Mucopolisacaridosis tipo III (sinónimos: síndrome de Sanfilippo, deficiencia de aN-acetilglucosaminidasa lisosomal - mucopolisacaridosis III A, deficiencia de acetil-CoA-a-glucosaminid-N-acetiltransferasa - mucopolisacaridosis III B, N-acetilglucosamina-6-sulfatasa - mucopolisacaridosis III C, deficiencia de sulfamidasa - mucopolisacaridosis III D).
 [
[ Patogenesia
La enfermedad está causada por mutaciones en cuatro genes diferentes: la aN-acetilglucosaminidasa lisosomal (mucopolisacaridosis III A), la acetil-CoA-a-glucosaminida-N-acetiltransferasa (mucopolisacaridosis III B), la N-acetilglucosamina-6-sulfatasa lisosomal (mucopolisacaridosis III C) y la sulfamidasa (mucopolisacaridosis III D). Todas estas enzimas participan en el metabolismo del heparán sulfato.
El gen de la heparán-N-sulfatasa (SGSH) se localiza en el brazo largo del cromosoma 17 (17q25.3). El 75,3 % de las mutaciones conocidas en el gen SGSH son puntuales. Se han descrito mutaciones frecuentes características de las poblaciones europeas: R74C (56 % en Polonia y 21 % en Alemania) y R245H (56 % en los Países Bajos).
La frecuencia de la mutación R74C es del 47,5% y la de la mutación R245H, del 7,5%. Las otras dos mutaciones descritas, delll35G y N389S, representan juntas el 21,7% de los alelos mutantes.
El gen de la aN-acetil-glucosaminidasa (NAGLU) se encuentra en el brazo largo del cromosoma 17 (17q21). El 69 % de las mutaciones encontradas en el gen NAGLU son mutaciones sin sentido y sin sentido, y el 26,3 % son pequeñas deleciones e inserciones. El gen de la acetil-CoA-γ-glucosaminida-N-acetiltransferasa (HGSNAT) se encuentra en el brazo corto del cromosoma 8 (8p11.1). Este gen se caracterizó en 2006 y, hasta la fecha, solo se han encontrado unas pocas mutaciones.
El gen de la N-acetilglucosamina-6-sulfatasa (GNS) se encuentra en el brazo largo del cromosoma 12 (12ql4). Hay 12 pacientes con mucopolisacaridosis IIID registrados en el mundo. Se han descrito cuatro mutaciones en el gen GNS.
En todos los subtipos de mucopolisacaridosis III, existe una alteración en la degradación del heparán sulfato, que forma parte de la estructura de las membranas celulares, incluidas las neuronales, lo cual se correlaciona con un proceso neurodegenerativo grave causado por atrofia cortical. La diarrea crónica se explica por la participación del sistema nervioso autónomo en el proceso patológico, junto con la disfunción de la mucosa intestinal. La hipoacusia neurosensorial probablemente se deba a tres causas: otitis frecuente, deformación de los huesecillos auditivos y anomalías del oído interno. La rigidez articular es resultado de la deformación de las metáfisis, mientras que el engrosamiento de la cápsula articular es secundario al depósito de glicosaminoglicanos y la fibrosis. Las diferencias intrasindrómicas en la gravedad de la enfermedad se deben exclusivamente a la actividad funcional residual de la enzima mutante: cuanto mayor es, más leve es la enfermedad.
Síntomas mucopolisacaridosis tipo 3
El polimorfismo clínico en el síndrome de Sanfilippo es menos pronunciado que en otros tipos de mucopolisacaridosis. Se caracteriza por una progresión lenta de la enfermedad y trastornos neurológicos graves con síntomas leves en órganos internos y otros sistemas.
Los primeros síntomas de la enfermedad suelen aparecer entre los 2 y los 6 años en niños con un desarrollo previo normal. Los síntomas más evidentes incluyen regresión del desarrollo psicomotor y del habla, trastornos psiquiátricos como el síndrome de hiperactividad, comportamiento autista o agresivo, trastornos del sueño y descuido y falta de atención.
Otros síntomas comunes son hirsutismo, vello grueso, hepatoesplenomegalia moderada, deformidad en valgo de las extremidades y cuello corto. El desarrollo de rasgos faciales toscos, como el gárgola, y deformidades esqueléticas como la disostosis múltiple se manifiestan débilmente en la mucopolisacaridosis III, en comparación con otros tipos de mucopolisacaridosis caracterizados por el fenotipo de Hurler. La estatura, por regla general, se corresponde con la edad, y la rigidez articular rara vez causa disfunción. La mayoría de los pacientes suelen desarrollar osteoporosis y osteomalacia. Trastornos esqueléticos secundarios: alto riesgo de fracturas patológicas. Los trastornos psiconeurológicos graves se observan con mayor frecuencia entre el sexto y el décimo año de vida y conducen a una marcada inadaptación social. La hipoacusia neurosensorial progresiva es inherente a todos los pacientes con formas graves y moderadas de la enfermedad. Se observan convulsiones en casi todos los pacientes a medida que la enfermedad progresa.
La enfermedad progresa rápidamente y la mayoría de los pacientes no sobreviven hasta los 20 años. La mucopolisacaridosis IIIA se considera el tipo más común y grave de este síndrome.
Diagnostico mucopolisacaridosis tipo 3
El diagnóstico de mucopolisacaridosis III se confirma determinando el nivel de excreción urinaria de glicosaminoglicanos y midiendo la actividad enzimática. En el caso de la mucopolisacaridosis III, la excreción total de glicosaminoglicanos en la orina aumenta y se observa hiperexcreción de heparán sulfato. La actividad de las enzimas lisosomales correspondientes a un subtipo específico de mucopolisacaridosis III se mide en leucocitos o en cultivos de fibroblastos cutáneos utilizando un sustrato fluorogénico artificial.
El diagnóstico prenatal se realiza midiendo la actividad enzimática en una biopsia de vellosidades coriónicas entre las semanas 9 y 11 de embarazo o determinando el espectro de glicosaminoglicanos en el líquido amniótico entre las semanas 20 y 22 de embarazo. En familias con genotipo conocido, el diagnóstico de ADN puede realizarse al inicio del embarazo.
¿Qué pruebas son necesarias?
Diagnóstico diferencial
El diagnóstico diferencial se realiza tanto dentro del grupo de las mucopolisacaridosis como con otras enfermedades de almacenamiento lisosomal: mucolipidosis, galactosialidosis, sialidosis, manosidosis, fucosidosis, gangliosidosis GM1.
¿A quién contactar?
Tratamiento mucopolisacaridosis tipo 3
Hasta la fecha, no se han desarrollado métodos terapéuticos eficaces para la mucopolisacaridosis III. Se indica tratamiento sintomático.
Использованная литература