Médico experto del artículo.

Nuevos artículos
Síndrome de Tolosa-Hunt
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
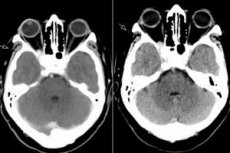
Síndrome de la fisura orbitaria superior, oftalmoplejía patológica: todo esto no es más que el síndrome de Tolosa Hunt, que consiste en una lesión de las estructuras de la fisura orbitaria superior. El proceso suele afectar los vasos orbitarios (arteriales y venosos), las fibras nerviosas (nervios oculomotor, troclear y abducens, así como la primera rama del nervio trigémino) y el seno cavernoso adyacente. Esta enfermedad puede clasificarse como una patología relativamente rara y de difícil diagnóstico. [ 1 ]
Epidemiología
El síndrome de Tolos Hunt se describió hace no mucho tiempo, hace unos 70 años. Fue estudiado por el neurólogo español E. Tolos. Unos años más tarde, el trabajo fue complementado por el oftalmólogo inglés W. Hunt. Los nombres de los médicos investigadores se convirtieron en la base del nombre del síndrome.
El síndrome de Tolosa Hunt se presenta por igual en hombres y mujeres. La patología suele ser unilateral y se observa con la misma frecuencia en el lado izquierdo o derecho. El síndrome bilateral es posible, pero solo se presenta en casos aislados.
La edad promedio de los afectados es de 50 años. En general, el síndrome de Tolosa Hunt se presenta entre los 15 y los 85 años. La mayoría de los pacientes pertenecen a la tercera edad: el desarrollo de la enfermedad se ve facilitado por múltiples trastornos cardiovasculares, así como por cambios tisulares relacionados con la edad.
El síntoma más común de la enfermedad es la manifestación de un ataque de migraña clásico: la persona experimenta una cefalea repentina, pulsátil y punzante en un lado, con irradiación a la cuenca del ojo. Dado que el síndrome de Tolosa Hunt carece de síntomas específicos típicos, esta patología suele denominarse "camaleón neurológico": el diagnóstico es complejo y requiere diferenciación de muchas otras enfermedades.
Los pacientes con síndrome de Tolosa Hunt se presentan periódicamente en diferentes países del mundo, sin características territoriales ni estacionales. La tasa de incidencia es de 0,3 a 1,5 casos por millón de habitantes. [ 2 ]
Causas Síndrome de Tolosa-Hunt
Durante la investigación de las causas del desarrollo del síndrome de Tolosa Hunt, los científicos descubrieron los siguientes hechos:
- En la mayoría de los casos, la enfermedad fue provocada por una inflamación inmune de la pared exterior del seno cavernoso;
- en algunos casos las causas fueron malformaciones vasculares, procesos tumorales en el cerebro (formas primarias y secundarias), paquimeningitis craneal localizada, miositis orbitaria, periarteritis nodosa y formación de trombos en el seno cavernoso;
- En aproximadamente el 30% de los pacientes no se puede determinar la causa del trastorno, por lo que se estableció el diagnóstico de síndrome de Tolosa Hunt idiopático.
Consideremos estas supuestas razones con más detalle.
- El desarrollo autoinmune del síndrome se asocia tanto a hipotermia como a enfermedades infecciosas recientes, así como a estrés intenso. La forma autoinmune de la enfermedad se caracteriza por: inicio agudo, curso recurrente y alta eficacia del tratamiento con glucocorticosteroides. Esta forma de la enfermedad afecta con mayor frecuencia a los hombres.
- Las malformaciones vasculares son frecuentes en la hipertensión arterial descompensada. Afecta con mayor frecuencia a las mujeres. La enfermedad comienza de forma aguda, con dolor moderado y prácticamente sin exoftalmos ni quemosis.
- Entre los procesos tumorales capaces de conducir al desarrollo del síndrome de Tolosa Hunt, los más frecuentes fueron los tumores cerebrales primarios, los tumores metastásicos con focos primarios en pulmón, bronquios, próstata o las metástasis de melanoma cutáneo.
- La paquimeningitis craneal localizada causa un inicio agudo del síndrome en ausencia de signos cerebrales y meníngeos generales, sin exoftalmos. El diagnóstico se confirma morfológicamente mediante biopsia.
- La miositis orbitaria cursa con un inicio subagudo, con dolor intenso y exoftalmos, quemosis pronunciada y visión doble.
- La trombosis del seno cavernoso causa oftalmoplejía total. El diagnóstico se confirma mediante resonancia magnética.
- La periarteritis nodular puede provocar el desarrollo del síndrome de Tolosa Hunt varios meses después del inicio de la enfermedad.
En la mayoría de los casos, el mecanismo autoinmune subyace a la formación de la patología, lo cual ha sido comprobado por numerosos especialistas. La naturaleza autoinmune se evidencia, en particular, por los siguientes factores:
- curso recurrente;
- trastornos dismúnicos;
- disociación proteína-célula en el líquido cefalorraquídeo y aumento de los niveles de citocinas proinflamatorias en el líquido cefalorraquídeo y el suero sanguíneo. [ 3 ]
Factores de riesgo
Los científicos aún no han determinado la causa exacta del síndrome de Tolosa Hunt. Sin embargo, han logrado identificar ciertos factores que influyen en el desarrollo de este trastorno:
- Predisposición genética a enfermedades autoinmunes en general. Si un familiar padeció o padece un trastorno autoinmune, otros familiares también podrían presentar patologías similares u otras con un mecanismo de desarrollo similar. Este factor aún es una suposición que requiere investigación y evidencia adicionales.
- Factores ambientales, incluidos los hábitos alimentarios, las condiciones ambientales, la calidad del agua, los riesgos industriales, etc.
- Situaciones de estrés severo, frecuentes shocks psicoemocionales y de estrés, cambios hormonales fuertes (incluido embarazo, menopausia, etc.).
- Enfermedades infecciosas crónicas de larga duración, incluidas hepatitis, infección por herpesvirus, citomegalovirus, etc.
- Hipotermia, radiación, otros irritantes fuertes y factores dañinos.
Patogenesia
El mecanismo etiológico del desarrollo del síndrome de Tolosa Hunt no se ha dilucidado por completo. Se atribuye un papel determinante a las reacciones autoinmunes. Muchos científicos asumen que las infecciones virales y microbianas, las situaciones de estrés y la radiación actúan únicamente como factor desencadenante. No existe evidencia sólida que respalde la relación entre la entrada de microorganismos patógenos en el organismo y el desarrollo del síndrome de Tolosa Hunt. Sin embargo, se sospecha la participación del citomegalovirus en el proceso autoinmune, que contribuye a la formación de granulomas. [ 4 ]
El esquema patogénico se debe a la aparición de un proceso inflamatorio granulomatoso local en la zona de la pared externa del seno cavernoso, sección infraclinoidea o supraclinoidea de la arteria carótida interna, lo que provoca su estrechamiento. El trastorno de la protección inmunitaria humoral y celular también desempeña un papel importante. La faceta humoral del síndrome se asocia con un aumento de la formación de anticuerpos citoplasmáticos antineutrófilos que actúan contra las enzimas proteinasa-3, mieloperoxidasa y una proteína de membrana específica capaz de unirse a endotoxinas. Presumiblemente, los anticuerpos citoplasmáticos estimulan a los neutrófilos existentes, lo que resulta en que ataquen los órganos diana; en particular, el proceso inflamatorio se desarrolla en la pared externa del seno cavernoso.
Los cambios celulares también influyen en el desarrollo del síndrome de Tolosa Hunt. Esto se demuestra por el predominio de linfocitos T, macrófagos y células plasmáticas en los granulomas.
Existe información sobre estructuras endoteliales altamente activas y citocinas antiinflamatorias, lo que indica una tendencia del proceso patológico a volverse crónico.
En casos aislados se observaron cambios necróticos focales en el área de la pared externa del seno cavernoso.
Síntomas Síndrome de Tolosa-Hunt
Los síntomas característicos del síndrome de Tolosa Hunt aparecen de forma repentina e inesperada. Los principales síntomas son los siguientes:
- Dolor intenso en la cuenca del ojo, extremadamente desagradable, perforante, que se extiende desde la región frontal hasta el arco superciliar, los ojos y más adelante por toda la cabeza.
- Visión doble, que se detecta tras la aparición del dolor. A la persona le resulta extremadamente difícil concentrarse visualmente y examinar cualquier objeto.
- El trastorno de la función motora del globo ocular, también conocido como oftalmoplejía, es predominantemente unilateral. Puede manifestarse en diversos grados, dependiendo de la gravedad del proceso patológico y la extensión de la lesión.
- Edema conjuntival.
- Desplazamiento anterior del globo ocular (exoftalmos, ojos “saltados”).
- Desviación del eje visual de un globo ocular hacia un lado, estrabismo, que es típico en caso de daño nervioso unilateral.
- Deterioro general del estado de salud, ligero aumento de temperatura, debilidad, irritabilidad.
El cuadro clínico progresa gradualmente, los síntomas cambian y empeoran, pero pueden desaparecer tan repentinamente como aparecieron. Sin embargo, si no se administra el tratamiento necesario, el síndrome de Tolosa Hunt vuelve a manifestarse con una recaída.
Los síntomas neurológicos se deben a la localización del proceso doloroso. El dolor se produce por la irritación de la primera rama del nervio trigémino, que discurre cerca del tronco del nervio oculomotor, y se observa en la zona de la órbita, la frente, la sien y la base de la nariz. La intensidad del dolor varía de moderada a intensa.
Es posible que se presenten síntomas atípicos, caracterizados por la ausencia de dolor. Esto se observa cuando la lesión se localiza antes de que el quinto par penetre en el seno cavernoso.
Los trastornos oculomotores generalmente se manifiestan como visión doble durante la mirada directa.
Si el proceso doloroso se localiza en la zona del vértice orbitario, suelen presentarse manifestaciones neurológicas en combinación con trastornos del analizador visual. Como resultado, se produce edema o atrofia del disco del nervio óptico, y a menudo se observa un escotoma central. Es posible que se presente exoftalmos (ojos saltones) y quemosis (edema conjuntival), cuya aparición se debe a cambios infiltrativos en el tejido retrobulbar y dificultades con el drenaje venoso orbitario.
Primeros signos
Dado que el síndrome de Tolosa Hunt no se ha estudiado lo suficiente hasta la fecha, los científicos continúan esclareciendo los posibles mecanismos de desarrollo de esta patología. Según los criterios de la Sociedad Neurológica Internacional, el diagnóstico del síndrome de Tolosa Hunt se justifica ante la presencia de un granuloma en la pared externa del seno cavernoso, detectado mediante resonancia magnética cerebral o biopsia.
La lista de signos que se aceptan como criterios diagnósticos del síndrome es la siguiente:
- dolor tipo "pinchazo" o "torsión" en la cuenca de un ojo con posterior desarrollo de parálisis muscular (oftalmoplejía);
- lesiones combinadas de los nervios oculomotores, la primera rama del nervio trigémino y el plexo nervioso periarterial;
- un aumento del cuadro clínico a lo largo de varios días (o en 1-2 semanas);
- la posibilidad de remisión espontánea (en algunos casos, con conservación residual de los defectos);
- la probabilidad de una recaída del síndrome, meses o años después;
- cuadro sistémico sin cambios, sin lesiones fuera del seno carotídeo;
- la presencia de un efecto positivo de la terapia con corticosteroides.
Existe otra lista diagnóstica similar de características propuesta en 2003. Según esta lista, se considera que el síndrome de Tolosa Hunt es el resultado de la proliferación de tejido granulomatoso en el seno cavernoso, la fisura orbitaria superior y la cavidad orbitaria:
- uno o más episodios de dolor unilateral en el área orbitaria que se resuelven sin tratamiento durante un par de semanas;
- daño del nervio craneal (III, IV o VI) en forma de paresia, presencia de granuloma confirmado mediante resonancia magnética o biopsia;
- la aparición de paresia simultáneamente con el síndrome doloroso, o dentro de los 14 días posteriores al mismo;
- desaparición de la paresia y del síndrome doloroso a los 3 días del inicio del tratamiento con corticosteroides.
Formas
En el síndrome de Tolosa Hunt, el lado izquierdo y el lado derecho se ven afectados con aproximadamente la misma frecuencia, por lo que la patología se divide en del lado izquierdo o del lado derecho.
La enfermedad suele ser unilateral. Solo en casos extremadamente raros se han observado lesiones bilaterales.
El cuadro clínico de la enfermedad puede evolucionar en las siguientes etapas:
- aguda o subaguda, que se produce después de una enfermedad infecciosa viral reciente, hipotermia, un fuerte aumento de la presión arterial, a veces sin una razón evidente;
- crónica recidivante, con aumento gradual de los síntomas y exacerbaciones periódicas.
Además, el síndrome de Tolosa Hunt puede ser:
- total, con daño de todos los nervios que pasan por la fisura orbitaria superior;
- incompleta, con afectación en el proceso patológico de los nervios pares VI, IV, III y rama I del par V en diversas combinaciones.
Respecto al seno, se pueden distinguir las formas anterior, media y posterior del síndrome de Tolosa Hunt.
Complicaciones y consecuencias
El síndrome de Tolosa Hunt se acompaña de dolor intenso, que conlleva pérdida de sueño y alteraciones emocionales y mentales. Los pacientes se vuelven irritables y emocionalmente inestables. Si no se realiza el tratamiento necesario, pueden aparecer trastornos neuróticos en este contexto: estados depresivos, neurastenia e hipocondría. La capacidad laboral se reduce significativamente y el paciente se retrae.
Un rasgo característico del síndrome de Tolosa Hunt es su evolución con recaídas, frecuente en enfermedades autoinmunes. La duración del período de remisión puede variar considerablemente: el indicador máximo registrado de duración asintomática fue de 11 años. Tras el tratamiento, el riesgo de recaídas se reduce significativamente. Si se producen exacerbaciones, estas son menos graves.
Diagnostico Síndrome de Tolosa-Hunt
A menudo resulta difícil para los médicos diagnosticar de inmediato el síndrome de Tolosa Hunt, ya que los síntomas son muy similares a las manifestaciones de otras enfermedades más comunes. En la mayoría de los casos, se requiere una consulta adicional con diversos especialistas: neurólogo, oftalmólogo, endocrinólogo, oncólogo, neurocirujano, etc.
En la primera etapa es necesario excluir enfermedades malignas, aneurismas, meningitis, etc.
En la mayoría de los casos, el síndrome de Tolosa Hunt se diagnostica por exclusión: el paciente se somete a una serie de pruebas para descartar otras enfermedades más probables. Se requieren las siguientes pruebas:
- imagen detallada de la sangre;
- estudio de la función hormonal de la glándula tiroides;
- estudio del nivel de proteína total en la sangre (para evaluar la calidad del metabolismo de las proteínas);
- análisis del líquido cefalorraquídeo.
- El diagnóstico instrumental implica la realización de los siguientes procedimientos diagnósticos:
- Resonancia magnética cerebral y de la región orbitaria, con y sin contraste;
- angiografía por resonancia magnética;
- angiografía por sustracción digital (angiografía por sustracción intravenosa);
- Tomografía computarizada cerebral y orbitaria con y sin contraste.
La resonancia magnética con gadolinio es la técnica de elección para la evaluación del síndrome de Thornton Heath (THS) y puede mostrar un agrandamiento y realce anormales del SC que se extiende a través de la fisura orbitaria superior hasta el ápex orbitario. Los hallazgos de la resonancia magnética, tanto en imágenes ponderadas en T1 como en T2, son extremadamente variables e inespecíficos. La resonancia magnética desempeña un papel fundamental en el diagnóstico y ayuda a descartar otras lesiones comunes asociadas con el SC, evitando la necesidad de procedimientos invasivos de alto riesgo como la biopsia de SC, la única forma de obtener la confirmación histopatológica de esta enfermedad.[ 5 ]
Estos estudios ayudan a identificar rastros de procesos inflamatorios en el seno cavernoso, la fisura orbitaria superior o el vértice orbitario. Los rastros de inflamación en la región orbitaria en imágenes transversales, en ausencia de parálisis de pares craneales, se consideran más benignos en términos de pronóstico.
A algunos pacientes con sospecha de síndrome de Tolosa Hunt se les aconseja someterse a una biopsia para descartar cáncer.
Diagnóstico diferencial
La práctica clínica indica que pueden presentarse síntomas similares en muchas patologías somáticas y neurológicas:
- en procesos inflamatorios microbianos, virales y fúngicos que afectan las meninges o la pared externa del seno cavernoso;
- en procesos tumorales en el cerebro y la órbita - por ejemplo, en adenoma hipofisario, craneofaringioma, neurinoma, meningioma del ala del hueso esfenoides, en metástasis cerebrales u orbitarias;
- en malformaciones vasculares, en particular, en aneurismas veno-arteriales, fístulas carótido-cavernosas, etc., así como en disecciones de ramas de la arteria carótida interna;
- para trombosis, formaciones quísticas del seno cavernoso, linfoma;
- para sarcoidosis, miositis orbitaria (músculos oculares), granulomatosis de Wegener (granulomatosis con poliangeítis), oftalmomigraña y algunas patologías sanguíneas.
El diagnóstico diferencial implica determinar la posibilidad de desarrollar todas las enfermedades mencionadas anteriormente, basándose en los resultados de una encuesta, examen y estudios de laboratorio e instrumentales.
Con mayor frecuencia, el síndrome de Tolosa Hunt debe distinguirse de las siguientes patologías:
- bloqueo del seno cavernoso por un trombo;
- Síndrome de Rochon-Duvignod;
- síndrome del espacio retroesfenoidal (síndrome de Jacot);
- síndrome de Raeder paratrigémino;
- polineuropatía craneal.
¿A quién contactar?
Tratamiento Síndrome de Tolosa-Hunt
El síndrome de Tolosa Hunt responde bien al tratamiento con un ciclo inmunosupresor de agentes hormonales corticosteroides. Estos fármacos pueden suprimir la respuesta agresiva del sistema inmunitario y su efecto dañino sobre los tejidos del organismo.
Los fármacos más comúnmente recetados son prednisolona, metilprednisolona, cortisona o fármacos alternativos que han demostrado efectos positivos en el tratamiento de patologías autoinmunes conocidas. Los beneficios de los esteroides probablemente se relacionen con el mecanismo antioxidante y/o la capacidad de dosis tan altas para reducir el edema y la isquemia subsiguiente en las zonas afectadas. [ 6 ]
Además de los corticosteroides, es adecuado el uso de analgésicos y anticonvulsivos. Es imprescindible el uso de multivitamínicos complejos.
Si sigue estrictamente todas las instrucciones y recomendaciones de su médico, los síntomas dolorosos del síndrome de Tolosa Hunt se alivian rápidamente: los pacientes notan una notable mejoría en su bienestar aproximadamente al segundo o tercer día. En la gran mayoría de los casos, se mantiene la capacidad laboral. [ 7 ]
Las dosis óptimas y la frecuencia de administración de medicamentos hormonales se determinan de forma individual. No existe un régimen de tratamiento generalmente aceptado, ya que es muy difícil organizar estudios controlados con placebo, lo cual se asocia a la baja prevalencia del síndrome. Con mayor frecuencia, se recomiendan dosis altas de corticosteroides, aunque se han observado casos de eficacia con dosis relativamente bajas (por ejemplo, el uso de prednisolona en cantidades inferiores a 0,5 mg/kg al día). Actualmente, la cantidad promedio de prednisolona utilizada en el síndrome de Tolosa-Hunt es de 1 a 2 mg/kg al día.
Plan de tratamiento aproximado:
- Metilprednisolona (Solu-Medron 1000 como infusión intravenosa por goteo con 250 ml de solución isotónica de cloruro de sodio y Panangin (10.0) diariamente durante cinco días;
- Mildronato para la normalización del metabolismo celular, 500 mg mediante inyección intravenosa diariamente durante 10 días;
- Neuromidina para mejorar la transmisión de impulsos a lo largo de las fibras neuromusculares, 20 mg por vía oral tres veces al día;
- Clonazepam para potenciar el efecto inhibidor sobre la transmisión de los impulsos nerviosos y la estimulación de los receptores de benzodiazepinas, 2 mg por vía oral, y/o Trileptal 150 mg por vía oral antes de acostarse.
Es posible prescribir un tratamiento prolongado con glucocorticosteroides utilizando dosis altas de prednisolona. [ 8 ]
Prevención
No es posible prevenir la aparición del síndrome de Tolosa Hunt. Esto se debe, al menos, a que las causas del trastorno aún no se han determinado con claridad. Si se detectan síntomas dolorosos, en particular dolor frecuente en la región frontal y las cuencas oculares, visión doble y debilitamiento de los músculos oculares, se debe contactar con el especialista adecuado lo antes posible para realizar un diagnóstico completo.
La prevención secundaria tiene como objetivo prevenir las recaídas en pacientes con síndrome de Tolosa Hunt ya diagnosticado. Los puntos importantes de las acciones preventivas son:
- Consultas médicas periódicas, procedimientos diagnósticos y seguimiento ambulatorio especializado;
- cursos periódicos de terapia con corticosteroides;
- Fortalecer y mantener un estado adecuado del sistema inmunológico.
Todas aquellas personas que estén enfermas deben tratar de evitar situaciones estresantes y tratar rápidamente cualquier proceso inflamatorio en el organismo.
Pronóstico
El pronóstico del síndrome de Tolosa Hunt se considera favorable. La respuesta al tratamiento con corticosteroides es favorable; los casos de remisión espontánea son frecuentes, aunque algunos pacientes experimentan efectos residuales en forma de deterioro de la función de los músculos oculares dañados. Si no se trata, la enfermedad puede recaer. En pacientes que han recibido tratamiento, se observan recaídas en aproximadamente el 35 % de los casos. [ 9 ]
Tras finalizar el tratamiento, la capacidad laboral suele restablecerse. Sin embargo, esto se aplica a una enfermedad correctamente diagnosticada, y no a otras patologías que se desarrollan bajo la apariencia del síndrome. [ 10 ]
La discapacidad se observa solo en casos excepcionales. Solo con exacerbaciones frecuentes documentadas se puede clasificar en el tercer grupo de discapacidad. En casos difíciles, se transfiere al paciente a trabajos ligeros, sin carga visual. Si el síndrome de Tolosa-Hunt es persistente y recurrente, no se recomienda conducir vehículos, debido al deterioro de la función motora ocular y a la diplopía.