Médico experto del artículo.

Nuevos artículos
Xeroderma pigmentoso
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
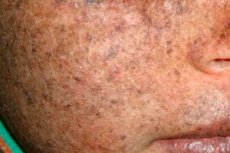
El xeroderma pigmentoso es un trastorno genético autosómico recesivo de la reparación del ADN. La enfermedad está causada por mutaciones en genes que participan en la reparación del ADN dañado por la radiación ultravioleta y otras sustancias tóxicas.
Esta enfermedad afecta con mayor frecuencia a los niños, también conocidos como "niños de la noche". Sus complicaciones frecuentes son el carcinoma basocelular y otras neoplasias malignas de la piel, el melanoma maligno metastásico y el carcinoma escamocelular.
 [ 1 ]
[ 1 ]
Patogenesia
La deficiencia o ausencia total de la enzima endonucleasa UV en las células del paciente (fibroblastos) desempeña un papel importante en el desarrollo de la enfermedad. Esta enzima es responsable de la reproducción del ADN dañado por los rayos ultravioleta. Según otras fuentes, la deficiencia de la enzima ADN polimerasa-1 desempeña un papel importante en el desarrollo de la enfermedad en algunos pacientes. La enfermedad se desarrolla con mayor frecuencia bajo la influencia de rayos con una longitud de onda de 280-310 nm. Se cree que la xerodermia pigmentaria se desarrolla debido a la entrada de diversas sustancias fotodinámicas en el organismo o al aumento de porfirinas en el entorno biológico humano.
En las primeras etapas de la enfermedad, se observa hiperqueratosis, atrofia del foco epidérmico, adelgazamiento de la capa de Malpighi, aumento de los gránulos de melanina en la capa basal celular e infiltración inflamatoria crónica en la capa superior de la dermis (principalmente alrededor de los vasos sanguíneos). Posteriormente, se detectan cambios degenerativos en el colágeno y las fibras elásticas, y en la etapa tumoral, se revelan signos característicos del cáncer de piel.
Síntomas xeroderma pigmentoso
Esta enfermedad afecta por igual a hombres y mujeres. Comienza en la primera infancia, en primavera o verano. La piel del paciente es especialmente sensible a la luz solar. Por lo tanto, la primera erupción, en forma de hiperpigmentación, similar a un lunar, aparece en las zonas de la piel expuestas a la luz solar. La erupción aumenta, el eritema se intensifica, se vuelve marrón oscuro y aparecen telangiectasias, angiomas y atrofia. Posteriormente, crecen papilomas y verrugas (principalmente en la piel de la cara y el cuello), y aparecen manchas y úlceras. Como resultado de la cicatrización de las úlceras, la nariz se adelgaza ("pico de pájaro") y el párpado se evierte. Los papilomas suelen convertirse en tumores malignos. La xerodermia pigmentada suele presentarse junto con el cáncer espinocelular y los melanosarcomas.
¿Qué te molesta?
Etapa
En el curso clínico del xeroderma pigmentoso se distinguen cinco estadios: inflamatorio (eritematoso), hiperpigmentación, atrofia, hiperqueratosis y tumores malignos.
Durante la fase inflamatoria (eritematosa), aparecen hinchazón, manchas rojas y, en ocasiones, ampollas y vesículas en las zonas de la piel expuestas a la luz solar (cara, cuello, parte superior del pecho, brazos y manos). Casi no se produce sarpullido en las zonas no expuestas a la luz solar.
En la hiperpigmentación, en lugar de las manchas rojas aparecen manchas hiperpigmentadas de color marrón claro, similares a lunares.
Con la atrofia, la piel se reseca, se adelgaza y se arruga. Se observan numerosas telangiectasias pequeñas y estrelladas, así como cicatrices con una superficie brillante, en la piel de los labios y la nariz. Debido a la atrofia y las cicatrices, se desarrolla microtomía (reducción de la abertura bucal), ectropión, adelgazamiento de las orejas y la nariz, y atresia de la abertura nasal y la boca. En el 80-85% de los pacientes, los ojos se ven afectados: se observan conjuntivitis, queratitis, daño a la córnea y las mucosas, y deterioro visual. Se observan discromías, telangiectasias, cambios hiperqueratósicos y tumores en la piel de los párpados.
Con hiperqueratosis, aparecen tumores verrugosos, papilomas, queratoacantomas, fibromas, angiofibriomas y otros tumores benignos en los focos patológicos descritos. Por ello, algunos científicos incluyen la xerodermia pigmentaria entre las enfermedades precancerosas.
Tras 10-15 años desde el inicio de la enfermedad, aparecen tumores malignos de la piel (basalioma, endotelioma, angiosarcoma) en focos y manchas pigmentarias atróficamente alterados. Estos tumores se destruyen rápidamente, metastatizan a órganos internos y provocan la muerte. En los órganos y tejidos internos de algunos pacientes, se observan cambios distróficos generales (sindactilia del segundo y tercer dedo del pie, distrofia dental, alopecia, etc.).
Formas
La forma neurológica del xeroderma pigmentoso se manifiesta en dos síndromes.
El síndrome de Reed se caracteriza por manifestaciones clínicas de xeroderma pigmentoso y microcefalia, idiopatía y crecimiento lento del sistema esquelético. En la forma neurológica de la enfermedad, la reparación del ADN es difícil y la radioterapia provoca un aumento de los principales focos patológicos.
El síndrome de Cocchione De Sinctis-X se caracteriza por los siguientes signos clínicos:
- desarrollo de signos clínicos de xeroderma pigmentoso en pieles particularmente fotosensibles;
- aparición temprana de tumores malignos;
- evolución simultánea de parálisis espástica, microcefalia y demencia;
- deformidades congénitas y enanismo;
- subdesarrollo de las gónadas;
- abortos frecuentes;
- transmisión recesiva por herencia.
Según algunos dermatólogos, el síndrome de Santis-Cacchione no es una enfermedad independiente, sino una manifestación clínica grave y plenamente expresada del xeroderma pigmentoso.
Formas genéticas
Tipos |
Gene |
Lugar |
Descripción |
Tipo A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentoso (XP) grupo A – forma clásica |
Tipo B, II, XPB |
XPB |
2t21 |
Grupo B de XP |
Tipo C, III, XPC |
XPC |
3p25 |
Grupo C de XP |
Tipo D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP Grupo D o Síndrome de De Sanctis-Cacchione |
Tipo E, V, XPE |
DDB2 |
11p12-p11 |
Grupo XP E |
Tipo F, VI, XPF |
ERCC4 |
16p13.3-p13.13 |
Grupo XP F |
Tipo G, VII, XPG |
RAD2ERCC5 |
13q33 |
Grupo G de XP y síndrome COFS (síndrome cerebro-óculo-faciosquelético) tipo 3 |
Tipo V, XPV |
POLACO |
6p21.1-p12 |
Variante Xeroderma Pigmentosa |
¿Qué es necesario examinar?
Cómo examinar?
¿A quién contactar?
Tratamiento xeroderma pigmentoso
Los antipalúdicos (delagyl, plaquenil, resoquin, etc.) protegen el ADN de los rayos ultravioleta, inhiben el proceso de despolimerización y reducen la sensibilidad (fotosensibilización) de la piel a la luz solar. Estos fármacos tienen propiedades antiinflamatorias e hiposensibilizadoras. Se recomienda realizar un tratamiento general junto con vitaminas (B1, B2, PP, B6, B12, A, E), antihistamínicos (tavegil, difenhidramina, suprastina) y desensibilizadores (tiosulfato de sodio, cloruro de calcio al 10 % por vía intravenosa, 10 ml).
Para el tratamiento local se utilizan cremas y ungüentos protectores solares.
En la forma tumoral de xerodermia pigmentaria, se utilizan métodos quirúrgicos, nitrógeno líquido y rayos láser. Para protegerse de la luz solar, debe usar ropa holgada, sombreros y guantes.
Pronóstico
La mayoría de los pacientes (2/3) mueren antes de cumplir los 15 años. Algunos pacientes pueden vivir hasta 40-50 años.
[ 20 ]