Médico experto del artículo.

Nuevos artículos
Linfomas cutáneos de células T
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
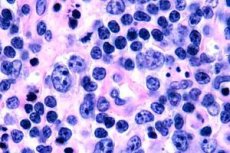
Los linfomas de células T se presentan con mayor frecuencia en personas mayores, aunque se han observado casos aislados incluso en niños. La incidencia es dos veces mayor en hombres que en mujeres. Los linfomas de células T son de naturaleza epidermotrópica.
Causas Linfomas cutáneos de células T
Las causas y la patogénesis de los linfomas cutáneos de células T no se comprenden por completo. Actualmente, la mayoría de los investigadores consideran al virus de la leucemia de células T humana tipo 1 (HTLV-1) I como el principal factor etiológico que inicia el desarrollo de linfomas malignos de células T en la piel. Asimismo, se analiza el papel de otros virus en el desarrollo del linfoma de células T: el virus de Epstein-Barr y el virus del herpes simple tipo 6. En pacientes con linfoma de células T, los virus se encuentran en la piel, la sangre periférica y las células de Langerhans. Se detectan anticuerpos contra el HTLV-I en muchos pacientes con micosis fungoide.
Un lugar importante en la patogenia de los linfomas de células T lo juegan los procesos inmunopatológicos en la piel, el principal de los cuales es la proliferación descontrolada de linfocitos clonales.
Las citocinas producidas por linfocitos, células epiteliales y células del sistema macrófago tienen efectos proinflamatorios y proliferativos (IL-1, responsable de la diferenciación linfocitaria; IL-2, factor de crecimiento de células T; IL-4 e IL-5, que aumentan la afluencia de eosinófilos a la lesión y su activación, etc.). Como resultado de la afluencia de linfocitos T a la lesión, se forman microabscesos de Pautrier. Simultáneamente con el aumento de la proliferación linfocitaria, se suprime la actividad de las células de defensa antitumorales: células asesinas naturales, linfocitos linfocitotóxicos, células dendríticas, en particular, células de Langerhans, así como citocinas (IL-7, IL-15, etc.), inhibidores del crecimiento tumoral. No se puede descartar el papel de los factores hereditarios. La presencia de casos familiares, la detección frecuente de algunos antígenos de histocompatibilidad (HLA B-5 y HLA B-35 - en los linfomas cutáneos altamente malignos, HLA A-10 - en los linfomas menos agresivos, HLA B-8 - en la forma eritrodérmica de la micosis fungoide) confirman la naturaleza hereditaria de la dermatosis.
Las observaciones clínicas indican una posible transformación de dermatosis crónicas a largo plazo (neurodermatitis, dermatitis atópica, psoriasis, etc.) en micosis fungoide. El factor clave es la persistencia prolongada de linfocitos en el foco inflamatorio, lo que altera la vigilancia inmunitaria y promueve la aparición de un clon de linfocitos malignos y, por consiguiente, el desarrollo de un proceso proliferativo maligno.
El impacto de factores físicos en el organismo, como la insolación, la radiación ionizante y las sustancias químicas, puede provocar la aparición de un clon de linfocitos “genotraumáticos” que tienen un efecto mutagénico sobre las células linfoides y el desarrollo de malignidad linfocitaria.
Por lo tanto, los linfomas de células T pueden considerarse una enfermedad multifactorial que comienza con la activación de los linfocitos bajo la influencia de diversos factores cancerígenos y genotraumatizantes, y la aparición de un clon dominante de células T. La gravedad del trastorno de la vigilancia inmunitaria y el clon de linfocitos malignos determinan las manifestaciones clínicas (elementos manchados, en placa o tumorales) de los linfomas de células T.
Patogenesia
En la etapa temprana de la micosis fungoide, se observa acantosis con procesos amplios, hiperplasia y compactación de los queratinocitos basales, degeneración vacuolar de algunas células basales, mitosis atípicas en diferentes capas de la epidermis y epidermotropismo del infiltrado con penetración de linfocitos en la epidermis. En la dermis, se observan pequeños infiltrados alrededor de los vasos, compuestos por células mononucleares individuales con núcleos hipercrómicos (células micóticas). En la segunda etapa, se observa un aumento de la gravedad del infiltrado dérmico y epidermotropismo de las células infiltradas, como resultado de lo cual los linfocitos malignos penetran en la epidermis, formando cúmulos en forma de microabscesos de Potrier. En la tercera etapa, la tumoral, se observa acantosis masiva y atrofia leve de la epidermis, así como un aumento de la infiltración epidérmica por linfocitos tumorales, que forman múltiples microabscesos de Potrier. El infiltrado masivo se localiza en todo el espesor de la dermis y cubre parte de la hipodermis. Se observan linfocitos en forma de blastos.
Linfoma cutáneo anaplásico de células T grandes
Se trata de un grupo de procesos linfoproliferativos caracterizados por la presencia de proliferaciones de linfocitos T CD30+ anaplásicos grandes, clonales y atípicos. Por lo general, se desarrolla de forma secundaria en la fase tumoral de la micosis fungoide o en el síndrome de Sézary, pero puede desarrollarse de forma independiente o con diseminación de linfomas sistémicos de este tipo. Clínicamente, estos linfomas corresponden a la denominada forma decapitada de la micosis fungoide, en forma de nódulos únicos o múltiples, generalmente agrupados.
Histológicamente, la proliferación ocupa casi toda la dermis con o sin epidermotropismo en el caso de atrofia epidérmica.
Citológicamente, las células tumorales pueden variar en tamaño y forma. Con base en estas propiedades, se distingue entre linfomas pleomórficos de células T de células medianas y grandes, con núcleos de diversas configuraciones irregulares: contorneados, multilobulados, con cromatina densa, un nucléolo bien definido y un citoplasma bastante abundante; inmunoblásticos, con núcleos grandes, redondos u ovalados, con carioplasma claro y un nucléolo central; y anaplásicos, con células muy grandes y de aspecto feo, con núcleos de configuración irregular y un citoplasma abundante. Fenotípicamente, todo este grupo pertenece a los linfomas de células T cooperadoras y pueden ser CD30+ o CD30-.
R. Willemze et al. (1994) demostraron que el linfoma CD30+ presenta una evolución más favorable. Genotípicamente, se detecta un reordenamiento clonal del receptor de linfocitos T.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
Síntomas Linfomas cutáneos de células T
La enfermedad más común dentro del grupo de linfomas cutáneos de células T es la micosis fungoide, que representa aproximadamente el 70% de los casos. Existen tres formas clínicas de la enfermedad: clásica, eritrodérmica y decapitada. Los linfomas de células T se caracterizan por el polimorfismo de erupciones en forma de manchas, placas y tumores.
La forma eritrodérmica de la micosis fungoide suele comenzar con picazón incontrolable, hinchazón, hiperemia generalizada y la aparición de lesiones eritematoescamosas en la piel del tronco y las extremidades, que tienden a fusionarse y desarrollar eritrodermia en un plazo de uno a dos meses. Casi todos los pacientes presentan hiperqueratosis palmoplantar y adelgazamiento difuso del cabello en toda la piel. Todos los grupos de ganglios linfáticos están muy aumentados de tamaño. Los ganglios linfáticos inguinales, femorales, axilares y cubitales aumentados de tamaño se palpan como "paquetes" de consistencia densa y elástica, no fusionados con los tejidos circundantes y sin dolor. El estado general se deteriora drásticamente: se presenta fiebre con una temperatura corporal de hasta 38-39 °C, sudores nocturnos, debilidad y pérdida de peso. Actualmente, muchos dermatólogos consideran que el síndrome de Sézary es la variante leucémica más rara de la forma eritrodérmica de la micosis fungoide.
Se observa una leucocitosis pronunciada en los linfocitogramas (células de Sézary). Estas células son células T cooperadoras malignas, cuyos núcleos presentan una superficie cerebriforme plegada con profundas invaginaciones en la membrana nuclear. Se observa un desenlace fatal después de 2 a 5 años, cuya causa frecuente es la patología cardiovascular y la intoxicación.
La forma decapitada de la micosis fungoide se caracteriza por el rápido desarrollo de lesiones tumorales en piel aparentemente sana, sin formación previa de placa a largo plazo. Esta forma se caracteriza por un alto grado de malignidad, lo que se considera una manifestación de linfosarcoma. El desenlace fatal se observa en un plazo de un año.
Etapa
La forma clásica de la micosis fungoide se caracteriza por tres estadios de desarrollo: eritematoescamoso, en placa y tumoral.
La primera etapa se asemeja al cuadro clínico de algunas dermatosis inflamatorias benignas: eccema, dermatitis seborreica y parapsoriasis en placas. En esta etapa de la enfermedad, se observan manchas de diversos tamaños, de color rosa intenso, rosa rojizo con un tinte púrpura, con contornos redondos u ovalados, con límites relativamente definidos, y descamación superficial en placas finas o con aspecto de salvado. Los elementos se localizan a menudo en diferentes áreas de la piel, con mayor frecuencia en el tronco y la cara. Su número aumenta gradualmente. Con el tiempo, el proceso puede adquirir el carácter de eritrodermia (etapa eritrodérmica). La erupción puede persistir durante años o desaparecer espontáneamente. A diferencia de las dermatosis inflamatorias benignas, los elementos de la erupción y el prurito en esta etapa son resistentes al tratamiento.
La fase de placa infiltrativa se desarrolla a lo largo de varios años. En lugar de las erupciones cutáneas previamente moteadas, aparecen placas de contornos redondos o irregulares, de color púrpura intenso, claramente delimitadas de la piel sana, densas y con una superficie escamosa. Su consistencia se asemeja a la de un cartón grueso. Algunas se resuelven espontáneamente, dejando zonas de hiperpigmentación marrón oscuro o atrofia (poiquilodermia). El prurito en esta fase es aún más intenso y doloroso, y se observa fiebre y pérdida de peso. También puede observarse linfadenopatía.
En la tercera etapa, la del tumor, aparecen tumores indoloros de consistencia densa y elástica, de color amarillo rojizo, que se desarrollan a partir de placas o surgen en piel aparentemente sana. La forma de los tumores es esférica o aplanada, a menudo similar a la de un hongo. Los tumores pueden aparecer en cualquier parte del cuerpo. Su número varía considerablemente, desde uno hasta decenas, y su tamaño, de 1 a 20 cm de diámetro. Cuando los tumores de larga duración se desintegran, se forman úlceras con bordes irregulares y fondo profundo, que alcanzan la fascia o el hueso. Los ganglios linfáticos, el bazo, el hígado y los pulmones son los más afectados. El estado general empeora, aparecen y aumentan los síntomas de intoxicación y se desarrolla debilidad. La esperanza de vida promedio de los pacientes con la forma clásica de micosis fungoide desde el momento del diagnóstico es de 5 a 10 años. La mortalidad suele deberse a enfermedades intercurrentes: neumonía, insuficiencia cardiovascular y amiloidosis. Subjetivamente, se percibe picazón y, cuando los tumores se desintegran, dolor en las zonas afectadas.
¿Qué es necesario examinar?
Tratamiento Linfomas cutáneos de células T
En la fase eritematoescamosa, los pacientes no requieren tratamiento antitumoral; se les prescriben corticosteroides tópicos (prednisolona, betametasona, derivados de la dexametasona), interferón alfa (3 millones de UI al día, luego 3 veces por semana durante 3-6 meses, según las manifestaciones clínicas o la eficacia del tratamiento), interferón gamma (100.000 UI al día durante 10 días, el ciclo se repite de 12 a 3 veces con un descanso de 10 días), terapia PUVA o terapia Re-PUVA. La eficacia de la terapia PUVA se basa en la formación selectiva de enlaces cruzados covalentes de psoralenos con el ADN en los linfocitos T cooperadores en proliferación, lo que inhibe su división. En la segunda etapa, además de los agentes mencionados, se utilizan corticosteroides sistémicos (30-40 mg de prednisolona al día durante 1,5-2 meses) y citostáticos (prospedina 100 mg al día, 4-5 inyecciones en total). La combinación de interferones con otros métodos terapéuticos (interferones + PUVA, interferones + citostáticos, interferones + retinoides aromáticos) tiene un efecto terapéutico más pronunciado.
En la etapa tumoral, el método principal es la poliquimioterapia. Se utiliza una combinación de vincristina (0,5-1 mg por vía intravenosa una vez al día, un total de 4-5 inyecciones) con prednisolona (40-60 mg al día por vía oral durante la quimioterapia), prospidina (100 mg al día, un total de 3 g) e interferones. Se recomiendan la fotodinámica, la terapia con haz de electrones y la fotoféresis (fotoquimioterapia extracorpórea).