Médico experto del artículo.

Nuevos artículos
Rabia infantil
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
La rabia, o hidrofobia, es una enfermedad viral aguda que se transmite por la mordedura de un animal infectado, con daño al sistema nervioso y desarrollo de encefalitis grave con desenlace fatal.
Epidemiología
El virus de la rabia, un flagelo para la salud pública desde la antigüedad, causa actualmente aproximadamente 59.000 muertes humanas al año, casi todas transmitidas por mordeduras de perro. Esto tiene un impacto económico significativo en los países en desarrollo, en particular en África y Asia, que son los que menos pueden soportar estas pérdidas. Sin embargo, a pesar de su tasa de mortalidad cercana al 100%, la rabia canina es una enfermedad totalmente prevenible, y los ejemplos históricos de erradicación de la rabia canina en el mundo desarrollado así lo demuestran. [ 1 ]
Causas rabia
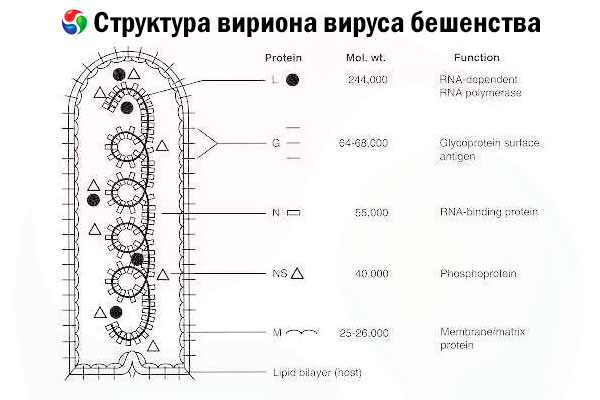
El agente causal es el virus de la rabia (RV), un virus ARN de cadena negativa de la familia de los rabdovirus, de un tamaño aproximado de 60 nm × 180 nm.
Consta de un núcleo proteico interno, o nucleocápside, que contiene ácido nucleico, y una membrana externa, una bicapa lipídica cubierta de espigas de glucoproteínas transmembrana. Presenta una estructura genómica modular relativamente simple y codifica cinco proteínas estructurales:
- ARN polimerasa dependiente de ARN (L),
- nucleoproteína (N),
- proteína fosforilada (P),
- proteína de matriz (M) y
- glicoproteína de superficie externa (G).
Las proteínas N, P y L, junto con el ARN genómico, forman el complejo ribonucleoproteico. G es el único antígeno del virus de la rubéola capaz de inducir la producción de anticuerpos neutralizantes del virus de la rubéola, que son los principales efectores inmunitarios contra la infección letal por este virus. Por otro lado, se ha demostrado que el complejo ribonucleoproteico es el principal antígeno del virus de la rubéola capaz de inducir la producción de linfocitos T CD4+, lo que puede potenciar la producción de anticuerpos neutralizantes del virus de la rubéola mediante el reconocimiento intraestructural del antígeno.[ 2 ] El complejo ribonucleoproteico puede desempeñar un papel importante en el establecimiento de la memoria inmunológica y la inmunidad a largo plazo.[ 3 ]
 [
[ Clasificación y tipos de antígenos
El género Lyssavirus incluye el virus de la rabia y otros virus de la rabia relacionados antigénica y genéticamente: los virus de Lagos, Mokola y Duvenhage (murciélago), así como dos supuestos subtipos de lyssavirus de murciélago europeos. Estudios de protección cruzada indican que los animales inmunizados con vacunas antirrábicas tradicionales podrían no estar completamente protegidos al ser expuestos a otros lyssavirus.
Los virus de la rabia se pueden clasificar como fijos (adaptados mediante el paso a animales o cultivos celulares) o callejeros (de tipo salvaje). El uso de anticuerpos monoclonales y la secuenciación genética para diferenciar los virus de la rabia callejeros ha ayudado a identificar variantes virales originadas en los principales reservorios de huéspedes a nivel mundial y a sugerir posibles fuentes de exposición humana cuando no existían antecedentes de una mordedura animal definitiva en el caso de un paciente.[ 8 ]
Patogenesia
El principal reservorio y fuente de infección entre los animales salvajes son los lobos, zorros, chacales y murciélagos, y entre los animales domésticos (perros y gatos, en raras ocasiones) los caballos, el ganado vacuno, los cerdos, las ratas, etc. La transmisión de la infección de persona a persona, aunque posible, es extremadamente rara. Se trata de una infección zoonótica típica. Las personas se infectan con rabia principalmente a través de los perros.
Tras la mordedura de un animal enfermo, el virus se multiplica en el tejido muscular del lugar de la mordedura y, tras alcanzar los extremos de los nervios periféricos sensoriales, se propaga centrípetamente hasta las neuronas motoras. El tiempo que tarda el virus en propagarse y afectar el cerebro depende del lugar de la mordedura. En mordeduras graves en la cabeza y la cara, el virus puede alcanzar el sistema nervioso central en 15-20 días, y en caso de daños menores en la piel del tronco y las extremidades, y consecuentemente una pequeña dosis del patógeno, el proceso de propagación del virus al sistema nervioso central puede demorarse varios meses o incluso entre 1 y 1,5 años. Una vez alcanzado el sistema nervioso central, el virus se fija en los tejidos del cerebro y la médula espinal, principalmente en las neuronas del bulbo raquídeo, el asta de Amón y la base del cerebro. En la médula espinal, las astas posteriores son las más afectadas. Desde el sistema nervioso central, el virus, mediante centrifugación a lo largo de los troncos nerviosos, llega a las glándulas salivales, donde se multiplica y se excreta con la saliva.
Conceptos sobre la patogénesis de la rabia
El virus de la rabia (VR) tiene un amplio espectro de hospedadores y puede infectar a casi todos los mamíferos. Aunque se han descrito diversas vías de transmisión, la infección natural se produce con mayor frecuencia por mordedura. Además de las mordeduras, el consumo de cadáveres infectados con el RV puede favorecer la infección por el virus de la rabia en zorros árticos, y se ha descubierto que el contacto del RV con las mucosas es otra posible vía de transmisión.[ 9 ] En circunstancias inusuales, como la liberación accidental de RV en forma de aerosol en un laboratorio o en cuevas habitadas por un gran número de murciélagos,[ 10 ] puede producirse la transmisión por aerosol.
Aún no está claro si el RV callejero y las cepas de RV adaptadas a ratones o a cultivos de tejidos se replican en el sitio de inoculación antes de entrar en el SNC. Si bien la infección intramuscular experimental de hámsteres o mapaches jóvenes con RV callejero reveló su replicación en células musculares estriadas antes de que el virus invadiera los axones de las neuronas motoras a través de las uniones neuromusculares,[ 11 ],[ 12 ] la infección intramuscular de ratones con RV CVS-24 adaptado a ratones mostró que el RV migra directamente al SNC sin replicación previa en el sitio de inoculación.[ 13 ] Una vez en las terminales de los axones amielínicos, el RV se transporta retrógradamente al cuerpo celular.
Hallazgos recientes sugieren que el transporte vesicular axónico puede representar una estrategia clave para el movimiento de viriones a larga distancia en los axones.[ 14 ] Se ha estimado que el RV migra dentro de los axones a una velocidad de 3 mm/h.[ 15 ] La infección luego se propaga a través de una cadena de neuronas conectadas por uniones sinápticas. Sin embargo, aún se desconoce el mecanismo exacto que promueve la propagación transsináptica. Después de infectar el cerebro, el virus se propaga centrífugamente al sistema nervioso periférico y autónomo en muchos órganos periféricos.[ 16 ] En la última etapa del ciclo de infección, el RV migra a las glándulas salivales; después de replicarse en las células acinares mucogénicas, se libera en la saliva y está listo para la transmisión al siguiente huésped.[ 17 ]
Con respecto a la patología inducida por el virus de la rabia, se ha propuesto la muerte celular apoptótica como un posible mecanismo patogénico en modelos experimentales de rabia de ratones infectados con una cepa fija de RV.[ 18 ] Un mecanismo patogénico que puede contribuir a la profunda disfunción del SNC característica de la rabia puede ser la función neuronal deteriorada. Se ha demostrado que la expresión génica se reduce notablemente en las neuronas infectadas con RV, lo que resulta en una supresión general de la síntesis de proteínas,[ 19 ] y varios estudios han mostrado una neurotransmisión deteriorada después de la infección por RV. Jiang demostró que la unión de un antagonista del receptor de acetilcolina a homogeneizados de cerebro de rata infectados se redujo en comparación con los controles.[ 20 ] También se observó una liberación y unión deterioradas de serotonina, un neurotransmisor involucrado en el control del ciclo del sueño, la percepción del dolor y el comportamiento, en el cerebro de rata infectado con RV. [ 21 ], [ 22 ] Además de afectar la neurotransmisión, la infección del ventrículo derecho también puede afectar los canales iónicos. Las células de neuroblastoma de ratón infectadas presentan una expresión funcional reducida de los canales de sodio dependientes de voltaje, lo que puede impedir los potenciales de acción y, en última instancia, conducir a un deterioro funcional. [ 23 ]
Además de la ausencia de lesiones patológicas graves en el SNC, la mayoría de los casos de rabia humana no provocan una respuesta inmunitaria de 7 a 10 días después del inicio de los signos clínicos. Estas profundas diferencias entre la patogénesis de la rabia y la de la mayoría de las otras infecciones virales o bacterianas del SNC se ven respaldadas además por el hecho de que la inmunosupresión es ineficaz o perjudicial para el resultado de la rabia.[ 24 ] El bajo nivel de respuesta inmunitaria que se observa a menudo en las víctimas de la rabia es desconcertante porque no se puede explicar por la baja inmunogenicidad de los antígenos del RV. De hecho, la proteína G del RV y la nucleocápside son potentes antígenos de células B y T cuando se administran por vía parenteral. [ 25 ] Una posible explicación para el bajo grado de respuesta inmunitaria contra el RV en humanos o animales con rabia puede ser que la infección del SNC por RV causa inmunosupresión, [ 26 ] y se ha propuesto que el RV utiliza una estrategia subversiva que incluye prevenir la apoptosis y destruir las células T invasoras. [ 27 ]
Las cepas atenuadas de RV adaptadas a células no neuronales difieren significativamente de las cepas patógenas de RV de origen local en su neuroinvasividad, es decir, su capacidad para invadir el SNC desde la periferia. En este sentido, las cepas de RV adaptadas a cultivos tisulares carecen o tienen una capacidad limitada para invadir el SNC desde la periferia, mientras que las cepas de RV de origen local o adaptadas a ratones, como la CVS-24, son altamente invasivas.[ 28 ] Los factores clave que intervienen en la neuroinvasión de RV incluyen la captación viral, el transporte axonal, la propagación transináptica y la tasa de replicación viral.
Hasta hace poco, nuestro conocimiento sobre la patogénesis del virus de la rabia era limitado y se basaba principalmente en estudios descriptivos de cepas de virus de la calle o infecciones experimentales con cepas atenuadas adaptadas en el laboratorio. La llegada de la tecnología de genética inversa nos ha permitido identificar los elementos virales que determinan el fenotipo patogénico del virus de la rabia y comprender mejor los mecanismos implicados en la patogénesis de la rabia.
Identificación de elementos virales que controlan la adquisición, diseminación y replicación del virus de la rabia
- Elementos virales implicados en la captura del virus
La infección por RV comienza con la unión del virus a un supuesto receptor celular. Aunque se han propuesto varias moléculas de la superficie de la membrana como receptores del RV, incluyendo el receptor nicotínico de acetilcolina,[ 29 ] la molécula de adhesión celular neuronal[ 30 ] y el receptor de neurotrofina de baja afinidad p75 NTR,[ 31 ] aún no está claro si estas moléculas realmente desempeñan un papel en el ciclo de vida del virus de la rabia. En este contexto, recientemente se ha demostrado que la interacción RV G–p75 NTR no es necesaria para la infección por RV de neuronas primarias.[ 32 ] Tras la unión al receptor, el RV se internaliza mediante endocitosis adsortiva o mediada por el receptor. [ 33 ] El entorno de bajo pH dentro del compartimento endosómico induce entonces cambios conformacionales en RV G que desencadenan la fusión de la membrana viral con la membrana endosómica, liberando así el RNP en el citoplasma. [ 34 ] En el caso de los virus, la proteína G del RV desempeña un papel fundamental en la captación viral, probablemente a través de interacciones con supuestos receptores celulares que facilitan una rápida captación. En este sentido, se ha demostrado que la patogenicidad de cepas de RV adaptadas a cultivos tisulares (p. ej., ERA, HEP y CVS-11) se correlaciona con la presencia de un determinante ubicado en el sitio antigénico III de la proteína G. [ 35 ] Una mutación Arg → Gln en la posición 333 de este sitio antigénico de la proteína G de ERA provocó un retraso de siete veces en la internalización de la variante Gln333 del RV en comparación con la variante silvestre. La mutación Asn194→Lys194 en RV G, que explica el resurgimiento del fenotipo patógeno, se asoció con una disminución significativa en el tiempo de internalización.[ 36 ] Además, los experimentos con RV quiméricos mostraron que el tiempo requerido para la internalización de los viriones de RV aumentó significativamente y la patogenicidad se redujo fuertemente después del reemplazo del gen G de la cepa SB RV altamente patógena, que se derivó de un clon de ADNc de la cepa RV-18 asociada a murciélagos derivada de plata,[ 37 ] con el de la cepa SN altamente atenuada, que se aisló de un clon de ADNc de la cepa de vacuna SAD B19 RV.[ 38 ] Juntos, estos datos respaldan la noción de que la cinética de la captación del virus, que es una función de RV G, es un determinante principal de la patogenicidad de RV.
- Elementos virales implicados en la propagación y transmisión de virus
Una propiedad única del virus de la rabia es su capacidad de propagarse entre células. La observación de que la variante Gln333 ERA pierde la actividad de fusión intercelular dependiente del pH in vitro [ 39 ] y muestra una capacidad de propagación intercelular considerablemente reducida [ 40 ] sugiere que la G del RV también desempeña un papel clave en la propagación intercelular y, por lo tanto, en la transmisión del virus, probablemente a través de su actividad fusiógena. Esta posibilidad se ve respaldada por el hallazgo de que la tasa de propagación del revertiente patógeno del RV SPBNGAK es casi el doble de alta que la determinada para la variante no patógena SPBNGA. Curiosamente, la mutación Asn 194 → Lys 194 en G SPBNGAK provocó un cambio en el umbral de pH para la fusión de membranas a un pH más alto, lo que respalda la hipótesis de que un umbral de pH más alto para la fusión de membranas se asocia con una mayor propagación del virus. [ 41 ]
Estudios de indicadores transneuronales de infección por RV en ratas [ 42 ] y monos rhesus [ 43 ] han demostrado que el virus de la rabia migra exclusivamente en dirección retrógrada en los axones. Aunque varias proteínas del RV están involucradas en mecanismos de transporte neuronal, RV G parece desempeñar un papel predominante en la propagación transneuronal de la infección por RV. Por ejemplo, mientras que la infección periférica con el virus de la anemia infecciosa equina (EIAV) pseudotipado con RV G resulta en la transferencia viral a la médula espinal, el mismo EIAV pseudotipado con el virus de la estomatitis vesicular G no logró ingresar al sistema nervioso. [ 44 ] Además, se encontró que la propagación viral del mutante ERA G Arg 333 → Gln 333 en el SNC estaba fuertemente reducida en comparación con el mutante de tipo salvaje, lo que sugiere además una función de RV G intacto en la propagación transsináptica. Sin embargo, la evidencia más convincente de un papel importante de RV G en el transporte trans-sináptico proviene de la infección intracraneal de ratones con un virus RV recombinante deficiente en G, que mostró que la infección permaneció restringida a las neuronas en el sitio de inoculación sin ninguna evidencia de propagación a neuronas secundarias.[ 45 ] Sin embargo, es probable que además de RV G, RV M también desempeñe un papel en la propagación del virus y por ende en el transporte trans-sináptico. A este respecto, se demostró que la propagación de la variante quimérica SN-BMBG RV, que contiene tanto M como G del SB altamente patógeno, fue significativamente mayor que la propagación de la variante quimérica SN-BG o SN-BM, que contienen la G y M del SB, respectivamente, lo que sugiere que la interacción óptima de M con G puede desempeñar un papel importante en la propagación del virus de célula a célula. [ 46 ] Dado que RV M favorece la gemación del virus, [ 47 ] es probable que la propagación más eficiente de la variante quimérica RV SN-BMBG se deba a la gemación óptima del virus en la membrana postsináptica.
Estudios recientes han demostrado que la interacción entre RV P y la cadena ligera de dineína vincula el RNP de RV al sistema de transporte de la célula huésped, facilitando así el transporte axonal retrógrado del virus.[ 48 ],[ 49 ] Sin embargo, la infección periférica de ratones adultos mostró que la eliminación del dominio de unión LC8 de RV P no impide la entrada del virus en el SNC, lo que sugiere que la proteína RV no está directamente involucrada en la propagación axonal retrógrada de RV.[ 50 ]
- Elementos virales que controlan la replicación viral
A diferencia de muchos otros virus, como el de la influenza, la patogenicidad del RV es inversamente proporcional a la tasa de síntesis de ARN viral y la producción de partículas virales infecciosas. La comparación de los niveles de ARNm viral y ARN genómico producidos por diferentes virus quiméricos sugiere que la transcripción y replicación del ARN viral están reguladas por múltiples factores, incluyendo la M del RV, que se ha identificado como un factor transactivo que media la transición de altos niveles iniciales de síntesis de ARNm a la síntesis de ARN genómico.[ 51 ] Además, la M de todos los rabdovirus es capaz de inhibir la expresión génica viral al unirse al RNP, lo que resulta en la formación de una estructura similar a una cadena principal altamente condensada que no puede soportar la síntesis de ARN.
Para identificar otros elementos virales que controlan la patogenicidad mediante la regulación de la replicación viral, las secuencias terminales 5' de la cepa SB altamente patógena se reemplazaron paso a paso con secuencias de la cepa de vacuna SN altamente atenuada, lo que resultó en los virus recombinantes SB2 (secuencia terminal [TS] + L), SB3 (TS + L + pseudogén [Ψ]), SB4 (TS + L + Ψ + G) y SB5 (TS + L + Ψ + G + M). La infección intramuscular con los virus parentales SB y SN y los RV quiméricos SB2, SB3, SB4 y SB5 provocó las tasas de mortalidad más altas en ratones infectados con SB y ninguna morbilidad o mortalidad en ratones infectados con SN. El reemplazo de TS, L y SB con los elementos correspondientes de SN resultó en una reducción modesta en la morbilidad y la mortalidad, y un intercambio adicional de G o G más M redujo fuertemente o abolió por completo la patogenicidad viral.
La caracterización fenotípica de estos RV de tipo silvestre y quiméricos en cultivo de tejidos reveló que la patogenicidad de un RV dado está inversamente correlacionada con su capacidad de replicarse en células neuronales. Si bien SB se replicó a niveles casi 1000 veces inferiores a SN, y la sustitución de TS, L y en SB por los niveles de SN tuvo poco efecto en la cinética de crecimiento viral, la sustitución adicional de la G o G más M de SB por los genes SN correspondientes resultó en un aumento de 1 logaritmo en la producción viral, lo que sugiere que la cinética de replicación del ARN viral, así como la producción de partículas virales, están controladas en gran medida por la proteína G del RV. Esta conclusión se sustenta en datos obtenidos con variantes de G del RV que difieren en un aminoácido en sus proteínas G. La variante patógena del virus de la rabia SPBNGAK 194 produjo un título viral en células NA que fue 1 log menor que el producido por la variante no patógena SPBNGAN 194, y el análisis de PCR en tiempo real mostró que las tasas de transcripción y replicación del ARN viral en células NA infectadas con SPBNGAK fueron 5 y 10 veces mayores que en las células NA infectadas con SPBNGAK.[ 52 ] Otra evidencia de una correlación inversa entre la patogenicidad y la tasa de síntesis de ARN viral y producción de partículas virales fue proporcionada por ratones infectados con virus recombinantes quiméricos en los que los genes G y M de la cepa SN atenuada fueron reemplazados por los de la cepa SB altamente patógena. Estos experimentos revelaron un aumento significativo en la patogenicidad de la cepa SN parental que transportaba RV G sobre la cepa SB patógena. La patogenicidad aumentó aún más cuando se introdujeron G y M de SB en SN.
La sustitución de G o M o ambas en SN con los genes correspondientes de SB se asoció con una disminución significativa en la tasa de producción de partículas virales, así como en la tasa de síntesis de ARN viral. Estos datos indican que tanto G como M desempeñan papeles importantes en la patogénesis de RV al regular la replicación viral. El hallazgo de que la sustitución de G o G más M en SN con G o G más M de SB resulta en una disminución moderada a fuerte en la transcripción y replicación del ARN viral, respectivamente, mientras que la sustitución de M sola en SN con M de SB resulta en un fuerte aumento en la transcripción y replicación del ARN viral, indica que RV G también tiene una importante función reguladora en la transcripción/replicación del ARN viral, ya sea sola o a través de la interacción con la proteína M. Se desconoce el mecanismo por el cual el gen RV G controla la síntesis de ARN viral. Ciertas secuencias de nucleótidos dentro de los genes RV G, como las que incluyen los codones para Arg333 y Lys 194, se han identificado como dianas para los miRNA celulares. Se ha demostrado que el reconocimiento de dianas por los miRNA celulares puede resultar en una regulación positiva o negativa de la replicación viral. [ 53 ] Las sustituciones Arg 333 → Glu 333 o Lys 194 → Ser 194 dentro de la secuencia del gen G del RV resultan en la abolición de las secuencias diana de los miRNA, lo que a su vez se asocia con un aumento significativo en la tasa de síntesis de ARN viral [Faber M, Thomas Jefferson University, PA, EE. UU., datos no publicados], lo que sugiere que los miRNA celulares del huésped también desempeñan un papel importante en la regulación de la replicación del RV, como se ha demostrado para otros virus de ARN, incluido el virus de la estomatitis vesicular y el VHC. [ 54 ], [ 55 ]
La regulación de la replicación viral parece ser uno de los mecanismos importantes involucrados en la patogénesis del virus de la rubéola. Para evadir la respuesta inmunitaria y preservar la integridad de la red neuronal, las cepas patógenas de virus de la rubéola, pero no las atenuadas, pueden regular su tasa de crecimiento. Una menor tasa de replicación probablemente beneficia a las cepas patógenas de virus de la rubéola, al preservar la estructura neuronal que estos virus utilizan para alcanzar el sistema nervioso central (SNC). Otra explicación para la menor tasa de replicación del virus de la rubéola patógeno es que, para evadir la detección temprana por parte del sistema inmunitario del huésped, el virus mantiene niveles mínimos de expresión de sus antígenos.
Relación entre la expresión de RV G, la apoptosis y la patogenicidad
Es bien sabido que las cepas del virus de la rabia callejera que son significativamente más patógenas que las cepas adaptadas a cultivos de tejidos expresan niveles muy limitados de G y no inducen apoptosis hasta tarde en el ciclo infeccioso, lo que sugiere que la patogenicidad de una cepa particular del virus está inversamente correlacionada con la expresión de G del RV y la capacidad de inducir apoptosis.[ 56 ] Se obtuvo evidencia directa de una correlación entre el nivel de expresión de G y el grado de apoptosis con el RV recombinante SPBNGA-GA, que portaba dos genes G idénticos y sobreexpresaba G del RV.[ 57 ] Los estudios morfológicos de cultivos neuronales infectados con este RV recombinante mostraron que la muerte celular aumentó significativamente en paralelo con la sobreexpresión de G del RV y que la apoptosis es el principal mecanismo implicado en la muerte mediada por G del RV. En particular, la disminución de la tinción de F-actina después de la infección con SPBNGA-GA es consistente con la despolimerización inducida por apoptosis de los filamentos de actina. Además, el número de núcleos TUNEL-positivos en neuronas infectadas con SPBNGA-GA aumentó significativamente en comparación con el de neuronas no infectadas e infectadas con SPBNGA. Sin embargo, el mecanismo por el cual el gen G de RV media el proceso de señalización apoptótica sigue siendo en gran parte desconocido. Se ha sugerido que la expresión de G de RV por encima de cierto umbral altera gravemente la membrana celular. Es muy probable que las células apoptóticas no se eliminen rápidamente en el SNC y, por lo tanto, sufran una necrosis secundaria. [ 58 ] Por otro lado, la infección por RV y, en particular, la sobreexpresión de la proteína G de RV pueden provocar piroptosis, una vía de muerte celular similar a la apoptosis que, a diferencia de la apoptosis, implica la activación de la caspasa 1 y, por lo tanto, provoca necrosis. [ 59 ] El grado de necrosis o piroptosis inducido por la infección por RV probablemente desempeña un papel fundamental en la inducción de la inmunidad antiviral. Mientras que las células apoptóticas mantienen la integridad de su membrana y no estimulan la respuesta inmune innata, las células necróticas se permeabilizan y secretan adyuvantes endógenos que pueden desencadenar una respuesta inmune innata robusta. [ 60 ]
Dado que el nivel de apoptosis/necrosis se correlaciona con la inmunogenicidad del RV, se ha sugerido que el efecto inmunoestimulante de las células apoptóticas/necróticas probablemente contribuya a la generación de una respuesta inmunitaria protectora. Por lo tanto, la regulación de la expresión de la proteína G del RV probablemente sea un factor importante en la patogénesis de la rabia, ya que facilita la supervivencia y la diseminación de variantes patógenas del RV en el sistema nervioso sin causar daño neuronal manifiesto y generando una respuesta inmunitaria protectora que prevendría la infección.
La expresión de RV G puede regularse a nivel de síntesis de ARN, a nivel postraduccional o a ambos. Se ha demostrado que los niveles de RV G expresados por diferentes variantes quiméricas de RV se reflejan en la tasa de síntesis de ARN viral, lo que sugiere que la regulación diferencial de la expresión de RV G por estas variantes resulta de variaciones en la tasa de transcripción de ARNm viral. Al igual que con las tasas de transcripción de ARN viral, la cantidad de RV G expresada por estas variantes se correlaciona inversamente con la patogenicidad viral. Por otro lado, la infección de cultivos neuronales primarios con la variante de RV menos patógena CVS-B2c resultó en niveles cuatro veces más altos de proteína G que la infección con la variante altamente patógena CVS-N2c, a pesar de la síntesis de niveles comparables de ARNm de G en ambas infecciones. Los experimentos de pulso-persecución mostraron que los niveles más altos de proteína G en neuronas infectadas con CVS-B2c fueron en gran parte el resultado de una menor tasa de degradación de la proteína G de CVS-B2c en comparación con la proteína G de CVS-N2c. Sin embargo, aún queda por dilucidar el mecanismo que conduce a la degradación proteolítica más rápida de la proteína G CVS-N2c.
Síntomas rabia
El período de incubación de la rabia es, en promedio, de 30 a 90 días. En caso de infección masiva a través de heridas extensas en la cabeza y la cara, puede acortarse a 12 días. En casos excepcionales, el período de incubación puede durar un año o más.
Hay un cambio estrictamente secuencial de tres períodos de la enfermedad: prodrómico, de excitación y de parálisis.
El período prodrómico comienza con la aparición de dolor o tirantez en el lugar de la picadura, así como dolor a lo largo de los nervios. En la zona de la cicatriz, puede haber sensación de ardor, picazón y, en ocasiones, enrojecimiento e hinchazón. El paciente experimenta malestar general, dolor de cabeza y náuseas. Se observan vómitos, aumento de la temperatura corporal a 37,5-38 °C y síntomas de un trastorno mental progresivo: aumento de la excitabilidad refleja, una inexplicable sensación de ansiedad, miedo y melancolía. A menudo, el paciente se muestra deprimido, inhibido, retraído, se niega a comer, duerme mal y se queja de pensamientos sombríos y pesadillas. El período prodrómico dura de 2 a 3 días, y en ocasiones puede extenderse hasta 7 días. Al final de este período, pueden presentarse ataques de ansiedad con dificultad respiratoria breve, sensación de opresión en el pecho, acompañada de taquicardia y aumento de la frecuencia respiratoria.
El período de excitación se caracteriza por la aparición de hidrofobia: al intentar beber, y al ver agua o recordarla, el paciente experimenta un espasmo convulsivo de la faringe y la laringe, durante el cual arroja la jarra de agua con un grito, extiende las manos temblorosas hacia adelante y echa la cabeza y el cuerpo hacia atrás. El cuello se estira, una mueca dolorosa distorsiona el rostro, que se vuelve azulado debido a un espasmo de los músculos respiratorios. Los ojos se salen de sus órbitas, expresan miedo, piden ayuda, las pupilas se dilatan y la respiración se dificulta. En el punto álgido del ataque, es posible un paro cardíaco y respiratorio. El ataque dura varios segundos, tras los cuales el estado del paciente parece mejorar. Posteriormente, pueden producirse espasmos de los músculos de la laringe y la faringe incluso por el movimiento del aire (aerofobia), la luz brillante (fotofobia) o una palabra fuerte (acusticofobia). Los ataques se acompañan de agitación psicomotora, durante la cual el paciente se comporta como un loco. La consciencia se nubla durante el ataque, pero se aclara en el período interictal. Durante el período de agitación, debido al aumento del tono del sistema nervioso simpático, los pacientes experimentan un aumento brusco de la salivación (sialorrea) con incapacidad para tragar saliva debido al espasmo de los músculos faríngeos. El paciente expulsa saliva. Algunos pacientes pueden desarrollar signos de meningismo e incluso opistótonos, y las convulsiones son frecuentes. En este caso, el líquido cefalorraquídeo puede no alterarse, pero en algunos pacientes, la concentración de proteínas puede aumentar y el número de células puede aumentar debido a los linfocitos.
Sin un tratamiento adecuado, los signos de deshidratación aumentan, los rasgos faciales se acentúan y el peso corporal disminuye. La temperatura corporal alcanza valores elevados. Es posible que se presenten convulsiones. La fase de excitación dura aproximadamente de 2 a 3 días, y rara vez de 4 a 5. El desenlace fatal suele ocurrir durante uno de los ataques. En raras ocasiones, el paciente sobrevive a la tercera etapa de la enfermedad.
Durante el período de parálisis, el paciente se calma. Los ataques de hidrofobia cesan, puede beber y tragar alimentos, y su consciencia es lúcida. Sin embargo, a pesar del aparente bienestar, pronto aparecen letargo, apatía y depresión, y parálisis de las extremidades, trastornos pélvicos y parálisis de los nervios craneales. La temperatura corporal sube a 42-43 °C, la presión arterial desciende y, al final del primer día, se produce la muerte por parálisis de los centros cardiovascular y respiratorio.
En la sangre periférica se observa leucocitosis neutrofílica, aumento de hemoglobina, eritrocitos y hematocrito.
¿Qué te molesta?
Formas
Clínicamente, se distinguen las formas típicas y atípicas. Las atípicas incluyen todos los casos sin excitación ni hidrofobia. Entre las formas atípicas se incluyen la bulbar, la cerebelosa, la meningoencefalítica, etc.
Diagnostico rabia
La detección del antígeno rábico, los anticuerpos, el ARN viral o el aislamiento del virus permiten el diagnóstico de la rabia. Dado que cualquier prueba individual puede ser negativa en un paciente con rabia, a veces son necesarias muestras seriadas de suero para la detección de anticuerpos antirrábicos, muestras de saliva para cultivo viral y biopsias cutáneas para la prueba de inmunofluorescencia directa del antígeno viral, especialmente cuando hay una alta sospecha de rabia.
Uno de los métodos más rápidos para diagnosticar la rabia ante mortem en humanos es realizar una prueba de inmunofluorescencia directa en una biopsia cutánea de la nuca para detectar el antígeno rábico. Esta prueba es el método más sensible y específico para detectar el antígeno rábico en la piel y otros tejidos frescos (p. ej., biopsia cerebral), aunque ocasionalmente los resultados pueden ser negativos en las primeras etapas de la enfermedad. Si no se dispone de tejido fresco, la digestión enzimática de tejidos fijados puede aumentar la reactividad de la prueba de inmunofluorescencia; sin embargo, la sensibilidad puede ser inaceptablemente baja.
El diagnóstico también puede establecerse aislando el virus de la saliva tras la inoculación de células de neuroblastoma o roedores de laboratorio; esto suele ser más eficaz durante las primeras 2-3 semanas de la enfermedad. La detección de anticuerpos neutralizantes del virus de la rabia, generalmente realizada mediante la prueba rápida de inhibición del foco fluorescente (RFFIT), en el suero de individuos no vacunados también es diagnóstica. La presencia de anticuerpos en el líquido cefalorraquídeo confirma el diagnóstico, pero pueden aparecer 2-3 días después que los anticuerpos séricos y, por lo tanto, pueden ser menos útiles en las primeras etapas de la enfermedad. Si bien la respuesta serológica tras la vacunación suele ser indistinguible de la respuesta serológica inducida por la enfermedad, la vacunación no suele producir anticuerpos contra el líquido cefalorraquídeo.
Solo se han documentado con precisión siete casos de "recuperación" de la rabia en los últimos 25 años. Si bien no se aisló el virus de la rabia en ninguno de los pacientes, los altos títulos de anticuerpos neutralizantes de la rabia en muestras de suero y la presencia de anticuerpos neutralizantes en el líquido cefalorraquídeo respaldaron firmemente el diagnóstico.
¿Qué es necesario examinar?
¿Qué pruebas son necesarias?
Diagnóstico diferencial
El diagnóstico de rabia humana suele basarse en datos epidemiológicos y clínicos, y se confirma en el laboratorio. El diagnóstico es sencillo si existen antecedentes de mordeduras de animales y se ha presentado el espectro completo de síntomas y signos. De lo contrario, es necesaria una evaluación cuidadosa pero rápida de las características epidemiológicas y clínicas de los casos menos típicos antes de realizar pruebas de laboratorio específicas. A todo paciente con signos o síntomas neurológicos o encefalitis inexplicable se le debe preguntar sobre la posibilidad de exposición a animales en zonas endémicas de rabia, dentro o fuera de su país de residencia. La falta de sospecha de rabia en varias muertes humanas recientes en Estados Unidos podría deberse a la falta de un historial detallado de exposición.
Al inicio de la enfermedad, la rabia puede imitar muchas enfermedades infecciosas y no infecciosas. Muchas otras encefalitis, como las causadas por herpesvirus y arbovirus, se asemejan a la rabia. Otras enfermedades infecciosas también pueden imitar la rabia, como el tétanos, la malaria cerebral, la rickettsiosis y la fiebre tifoidea. Entre las enfermedades infecciosas paralíticas que pueden confundirse con la rabia se encuentran la poliomielitis, el botulismo y la encefalitis por herpes simio B.
Las enfermedades no infecciosas que pueden confundirse con la rabia incluyen diversos síndromes neurológicos, en especial la polineuropatía inflamatoria aguda (síndrome de Guillain-Barré), así como la encefalomielitis alérgica posvacunal secundaria a la vacunación antirrábica del tejido nervioso, la intoxicación por drogas o intoxicación, la abstinencia alcohólica, la porfiria aguda y la histeria rábica. El síndrome de Guillain-Barré puede confundirse con la rabia paralítica, y viceversa.
¿A quién contactar?
Tratamiento rabia
No se ha desarrollado un tratamiento para la rabia. La administración de grandes dosis de inmunoglobulina antirrábica específica e interferón leucocitario resulta ineficaz. Se administra tratamiento sintomático para aliviar el sufrimiento del paciente. Para ello, se le coloca en una sala o box separado, donde se le establece un régimen de protección que limita la influencia del entorno externo (reducción de ruido, luz brillante, ventilación). Para reducir la excitabilidad del sistema nervioso central, se recetan somníferos, anticonvulsivos y analgésicos. Se normaliza el equilibrio hídrico.
En la fase paralítica, se prescriben fármacos que estimulan la actividad de los sistemas cardiovascular y respiratorio. Se recomienda el uso de oxigenación hiperbárica, hipotermia cerebral y respiración mecánica controlada, con curación completa del paciente. Sin embargo, todos los métodos de tratamiento son prácticamente ineficaces. En el mejor de los casos, es posible prolongar la vida del paciente varios meses. Un pronóstico desfavorable está determinado por la gravedad del daño al tronco encefálico, con la destrucción de centros vitales.
Prevención
El desarrollo de la primera vacuna antirrábica por Pasteur en 1885 marcó el comienzo de una era de control de la rabia mucho más eficaz. Hoy en día, a pesar de la tasa de mortalidad cercana al 100% en humanos por rabia, la enfermedad es completamente prevenible mediante la vacunación pre y/o postexposición. Si bien Pasteur y sus colegas iniciaron la vacunación de perros privados en París, la primera vacunación masiva de perros se llevó a cabo a principios de la década de 1920 en Japón, lo que marcó el primer programa nacional importante de control de la rabia. La vacunación oral de animales salvajes, desarrollada por primera vez en la década de 1970, ha demostrado desde entonces repetidamente que controla eficazmente la enfermedad en los principales huéspedes terrestres, como zorros, mapaches y zorrillos.[ 68 ] La vacunación sostenida contra la rabia de las poblaciones de animales reservorio con tasas de cobertura del 70% o superiores eventualmente eliminará el virus de la rabia de las especies reservorio y evitará la propagación del virus a huéspedes incidentales.[ 69 ]
Los datos filogenéticos indican que los lyssavirus infectaron a los murciélagos mucho antes de infectar a los mamíferos terrestres, y la mayoría de los lyssavirus, incluido el RABV, aún circulan en diversas especies de murciélagos en todo el mundo.[ 70 ] Sin embargo, los métodos efectivos para prevenir la transmisión del RABV entre murciélagos siguen siendo difíciles de alcanzar, lo que descarta la posibilidad de erradicación completa de la rabia en este momento. No obstante, incluso después de la exposición al RABV a través de la mordedura de un mamífero infectado con rabia, la profilaxis posexposición segura y efectiva (PEP, que incluye limpieza de heridas, inmunoglobulina antirrábica y vacunación antirrábica) puede proteger a los humanos de la infección por rabia si el tratamiento se administra con prontitud y de acuerdo con las recomendaciones de la Organización Mundial de la Salud (OMS).
Estos dos métodos para prevenir muertes humanas —uno basado en la vacunación de las personas expuestas y el otro en la vacunación de suficientes perros para romper el ciclo de transmisión en el origen— son los pilares de un enfoque de "una sola salud" para la prevención y el control de la rabia canina. Estos dos métodos se consideraron como alternativas independientes: la Estrategia A, basada en proporcionar PPE a las personas, y la Estrategia B, basada en la vacunación de perros; o como componentes de una Estrategia combinada A + B en un análisis de los costos probables de las estrategias alternativas.[ 71 ]
Países como Tailandia han tenido un enorme éxito en la prevención de muertes humanas mediante el uso de PEP, pero también han encontrado una creciente demanda y costos asociados asociados con el uso de PEP solo. [ 72 ] Por ejemplo, en comparación con la situación en 1991, cuatro veces más personas (más de 400.000) necesitaron PEP en 2003. Datos recientes muestran que la República Popular China, que vacuna a 15 millones de personas por año después de una posible exposición a la rabia, gasta alrededor de US$650 millones por año solo en PEP. [ 73 ]
Un enfoque mucho más sostenible consiste en prevenir la propagación de la infección en su origen, en la población animal, y a la vez aumentar el acceso a la profilaxis post exposición (PEP) para los pacientes humanos expuestos cuando sea necesario. Cuando existe voluntad política y financiación adecuada para controlar la rabia canina, se pueden eliminar, y de hecho se han eliminado, las muertes. El uso generalizado de la vacunación canina ha llevado a la eliminación de la rabia canina en varios países, como Malasia en 1954, [ 74 ] Japón en 1956, Taiwán en 1961, Singapur y, en particular, en toda Europa Occidental (revisado en Rupprecht et al., King et al., y Gongal y Wright). [ 75 ]
Использованная литература