Médico experto del artículo.

Nuevos artículos
Queratodermia: causas, síntomas, diagnóstico, tratamiento
Último revisado: 07.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
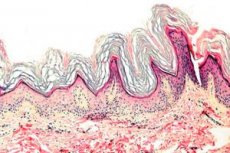
La queratodermia es un grupo de dermatosis que se caracterizan por una alteración del proceso de queratinización (formación excesiva de tejido córneo, principalmente en las palmas de las manos y las plantas de los pies).
Las causas y la patogénesis de la enfermedad no se han dilucidado por completo. Las investigaciones han establecido que las queratodermias son causadas por mutaciones en los genes que codifican la queratina 6, 9 y 16. La deficiencia de vitamina A, las disfunciones hormonales, principalmente de las glándulas sexuales, y las infecciones bacterianas y virales son de gran importancia en la patogénesis. Son uno de los síntomas de enfermedades hereditarias y tumores de órganos internos (queratodermias parapsoriásicas).
Síntomas. Se distingue entre queratodermia difusa (queratodermia de Unna-Tost, queratodermia de Meleda, queratodermia de Papillon-Lefevre, queratodermia mutilante y síndromes que incluyen la queratodermia difusa como uno de los síntomas principales) y focal (queratodermia manchada diseminada de Fischer-Buschke, acroqueratoelastoidosis de Kosti, queratodermia limitada de Bruhauer-Franzesthesti, queratodermia lineal de Fuchs, etc.).
La queratodermia de Winy-Tost (sinónimos: ictiosis congénita de palmas y plantas, síndrome de Winy-Tost) se transmite de forma autosómica dominante. Se observa una queratinización excesiva y difusa de la piel de palmas y plantas (a veces solo de plantas), que se desarrolla durante los dos primeros años de vida. El proceso patológico cutáneo comienza con un ligero engrosamiento de la piel de palmas y plantas, en forma de una franja eritematosa de color lívido en el borde de la piel sana. Con el tiempo, aparecen capas córneas lisas y amarillentas en su superficie. La lesión rara vez se extiende al dorso de las muñecas o los dedos. En algunos pacientes, pueden formarse grietas superficiales o profundas y se observa hiperhidrosis local. En el paciente observado por el autor, el tío materno, el hermano y el hijo padecían queratodermia de Winy-Tost.
Se describen casos de daños en las uñas (engrosamiento), dientes y cabello en la queratodermia de Winy-Tost en combinación con diversas anomalías esqueléticas y patologías de los órganos internos, sistemas nervioso y endocrino.
Histopatología. El examen histológico revela hiperqueratosis marcada, granulosis, acantosis y pequeños infiltrados inflamatorios en la dermis superior. Diagnóstico diferencial. La enfermedad debe diferenciarse de otros tipos de queratodermia.
La queratodermia de Meleda (sinónimos: enfermedad de Meleda, acroqueratoma progresivo congénito, queratosis palmoplantar transgradiente de Siemens, queratosis palmoplantar progresiva hereditaria de Kogoy) se hereda de forma autosómica recesiva. Esta forma de queratodermia se caracteriza por capas córneas gruesas de color amarillo-marrón con grietas profundas. Un borde violeta-morado de varios milímetros de ancho es visible a lo largo de los bordes de la lesión. El proceso suele extenderse al dorso de las manos y los pies, los antebrazos y las espinillas. La mayoría de los pacientes experimentan hiperhidrosis local. En este sentido, la superficie de las palmas de las manos y las plantas de los pies se humedece ligeramente y se cubre de puntos negros (conductos de las glándulas sudoríparas).
La enfermedad puede desarrollarse entre los 15 y los 20 años. Las uñas se engrosan y se deforman.
Histopatología. El examen histológico revela hiperqueratosis, en ocasiones acantosis, y un infiltrado inflamatorio crónico en la dermis papilar.
Diagnóstico diferencial. La queratodermia de Melela debe distinguirse de la queratodermia de Unna-Tost.
La queratodermia de Papillon-Lefevre (sinónimo: hiperqueratosis palmoplantar con periodontitis) se hereda de forma autosómica recesiva.
La enfermedad se manifiesta entre el segundo y tercer año de vida. El cuadro clínico es similar al de la enfermedad de Melela. Además, son característicos los cambios en la dentición (anomalías en la erupción de los dientes de leche y permanentes con desarrollo de caries, gingivitis, periodontosis de rápida progresión con pérdida prematura de piezas dentales).
Histopatología. El examen histológico revela engrosamiento de todas las capas de la epidermis, especialmente la capa córnea, y cúmulos celulares insignificantes de linfocitos e histiocitos en la dermis.
Diagnóstico diferencial. La enfermedad debe distinguirse de otras queratodermias. Un rasgo distintivo importante es la patología dental característica, que no se encuentra en otras formas de queratodermias difusas hereditarias.
La queratodermia mutilante (sinónimos: síndrome de Fonwinkel, queratoma mutilante hereditario) es un tipo de queratodermia difusa que se hereda de forma autosómica dominante. Se desarrolla durante el segundo año de vida y se caracteriza por depósitos córneos difusos en la piel de las palmas y plantas, con hiperhidrosis. Con el tiempo, se forman surcos en forma de cordón en los dedos, lo que provoca contracturas y amputación espontánea de los mismos. La queratosis folicular se manifiesta en el dorso de las manos, así como en la zona de las articulaciones del codo y la rodilla. Las láminas ungueales presentan alteraciones (a menudo como cristales de reloj). Se han descrito casos de hipogonadismo, alopecia rubí, pérdida auditiva y paquioniquia.
Histopatología. El examen histológico revela hiperqueratosis grave, granulosis, acantosis y pequeños infiltrados inflamatorios en la dermis, compuestos por linfocitos e histiocitos.
Diagnóstico diferencial. Al diferenciar la queratodermia mutilante de otras formas de queratodermia difusa, debe considerarse en primer lugar el efecto de mutilación, que no es característico de otras formas. Al realizar el diagnóstico diferencial de todas las formas de queratodermia difusa, es necesario recordar que puede ser uno de los síntomas principales de diversos síndromes hereditarios.
Tratamiento. La neotigazona está indicada en el tratamiento general de la queratodermia. La dosis del fármaco depende de la gravedad del proceso y es de 0,3 a 1 mg/kg de peso corporal del paciente. En ausencia de neotigazona, se recomienda vitamina A en dosis de 100 a 300.000 mg al día durante un periodo prolongado. El tratamiento externo consiste en el uso de ungüentos con retinoides aromáticos, agentes queratolíticos y esteroides.
 [
[ ¿Qué te molesta?
¿Qué es necesario examinar?
Cómo examinar?