Médico experto del artículo.

Nuevos artículos
Síndrome hemofagocítico en niños: primario, secundario
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
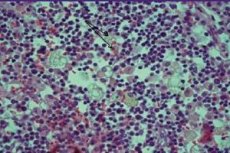
El síndrome hemofagocítico, también conocido como linfohistiocitosis hemofagocítica, es una enfermedad rara y difícil de definir. Esta grave enfermedad se asocia con la aparición de insuficiencia orgánica como resultado de la activación incontrolada del componente efector de la defensa inmunitaria celular.
En el síndrome hemofagocítico, se produce un fuerte aumento de la funcionalidad de los linfocitos T tóxicos y los macrófagos, lo que conduce a la producción de numerosas citocinas antiinflamatorias. A su vez, este proceso conlleva una intensa reacción inflamatoria sistémica y una disfunción a gran escala de muchos órganos.
Causas síndrome hemofagocítico
El síndrome hemofagocítico suele ser primario, es decir, de naturaleza hereditaria, como resultado de un trastorno genético en el funcionamiento de los macrófagos.
El síndrome hemofagocítico secundario también se denomina adquirido: se asocia a diversas patologías infecciosas, procesos tumorales, enfermedades autoinmunes y trastornos metabólicos congénitos.
En la variante clásica del síndrome hemofagocítico hereditario, los niños suelen ser ingresados en la unidad de cuidados intensivos (UCI) de hospitales especializados en enfermedades infecciosas, donde se les diagnostican complicaciones sépticas o infección intrauterina generalizada. El diagnóstico de síndrome hemofagocítico suele establecerse tras un desenlace fatal.
Sin embargo, incluso enfermedades infecciosas aparentemente comunes de origen viral o microbiano pueden causar una complicación como el síndrome hemofagocítico potencialmente mortal.
El síndrome hemofagocítico en adultos es casi siempre secundario: la patología se desarrolla con mayor frecuencia en el contexto de enfermedades linfoproliferativas e infecciones crónicas por VEB.
El síndrome hemofagocítico en niños puede ser primario o secundario, como resultado de enfermedades infecciosas previas (varicela, meningoencefalitis, etc.).
Síntomas síndrome hemofagocítico
Los síntomas del síndrome se describieron por primera vez a mediados del siglo pasado. Se identificaron los siguientes síntomas característicos:
- condición febril estable;
- disminución del nivel de sustancias hematopoyéticas;
- agrandamiento del hígado y del bazo;
- síndrome hemorrágico expresivo.
Los pacientes pueden presentar insuficiencia hepática, niveles elevados de ferritina y transaminasas, hallazgos neurológicos claros con disfunción del SNC, niveles elevados de triglicéridos séricos, coagulación sanguínea acelerada y coagulopatía.
A menudo, los pacientes presentan ganglios linfáticos agrandados, erupciones cutáneas, coloración amarillenta de la esclerótica, la piel y las membranas mucosas, así como hinchazón.
El parénquima del bazo, los capilares hepáticos sinusoidales, los senos linfáticos, la médula ósea y el sistema nervioso central se caracterizan por una infiltración difusa de macrófagos activos, en un contexto de síntomas hemofagocíticos. Se produce depleción del tejido linfoide. El examen hepático revela lesiones típicas de la forma crónica de inflamación persistente.
Formas
Existen dos formas clínicas que al principio son bastante difíciles de diferenciar.
- Linfohistiocitosis hemofagocítica primaria, que es una patología autosómica recesiva, en cuyo desarrollo tiene importancia primordial la mutación del gen de la perforina.
- Una forma secundaria de linfohistiocitosis hemofagocítica que se desarrolla como resultado de una actividad inmune excesiva de la cadena fagocítica mononuclear.
Complicaciones y consecuencias
- Adición de infección con intoxicación subsiguiente. Esta complicación se caracteriza por la pérdida gradual de la función de los principales órganos y sistemas, fiebre y agotamiento del paciente.
- Transformación maligna de células. La malignidad suele representar el desarrollo de linfoma, leucemia y otras enfermedades malignas.
- Las patologías autoinmunes son una especie de reacción agresiva de las defensas inmunitarias del propio paciente.
- Disminución persistente de la inmunidad con desarrollo de un estado de inmunodeficiencia.
- Insuficiencia de la función renal y hepática.
- Hemorragias internas, hemorragias.
- Muerte de un paciente por disfunción orgánica total o por complicaciones sépticas.
Diagnostico síndrome hemofagocítico
Si no hay antecedentes familiares, es muy difícil determinar la naturaleza primaria o secundaria del síndrome hemofagocítico. Para un diagnóstico preciso, es necesario realizar la diferenciación histológica de la hemofagocitosis.
Muchas enfermedades son difíciles de diagnosticar utilizando únicamente información obtenida de biopsias de tejidos: ganglios linfáticos, hígado y médula ósea.
Los estudios inmunológicos que permiten observar la función suprimida de las estructuras de las células NK y un aumento en el contenido del receptor de interleucina-2 no sirven como base para el diagnóstico. Además, se tienen en cuenta las características del cuadro clínico, el daño y la disfunción del sistema nervioso central, y los cambios en la composición sanguínea del paciente.
El punto final para realizar un diagnóstico son los datos del análisis genético molecular.
Diagnóstico diferencial
Diferenciar la enfermedad es extremadamente difícil, y el abordaje debe determinarse según la edad del paciente. En pediatría, es importante detectar las formas genéticas del síndrome hemofagocítico lo antes posible, analizando todos los factores que puedan indicar una patología hereditaria.
Por lo tanto, el rápido desarrollo del síndrome durante los primeros 12 meses de vida, con antecedentes familiares sin complicaciones, es característico de la forma primaria del síndrome hemofagocítico. La expresión de perforina en las estructuras de las células NK, detectada mediante citofluorometría de flujo y pruebas genéticas moleculares de perforina, ayuda a establecer el diagnóstico correcto en aproximadamente el 30 % de los casos de síndrome hemofagocítico hereditario. La aparición simultánea de la enfermedad en un contexto de albinismo se detecta en los siguientes síndromes:
Si la herencia es ligada al cromosoma X, es decir, cuando la enfermedad se desarrolla en varones emparentados por el lado materno, lo más probable es la presencia de síndrome linfoproliferativo autoinmune.
En el síndrome hemofagocítico secundario, lo principal es detectar oportunamente los tumores malignos, que son la causa más frecuente del síndrome en la edad adulta.
¿A quién contactar?
Tratamiento síndrome hemofagocítico
El tratamiento del síndrome hemofagocítico es bastante complejo: el éxito de dicho tratamiento depende en gran medida de la edad del paciente y de qué tan oportunamente se detectó la enfermedad.
Los regímenes terapéuticos para el síndrome hemofagocítico incluyen el uso de glucocorticosteroides (dexametasona) y citostáticos (etopósido, ciclosporina A). Los citostáticos se prescriben para suprimir la acción proinflamatoria de los fagocitos, con posterior trasplante alogénico de células madre.
Aún no se ha determinado un régimen de tratamiento único para el síndrome hemofagocítico. El tratamiento etiotrópico se considera insuficiente para combatir el síndrome, y el uso de inmunosupresores puede afectar negativamente la evolución del proceso viral-bacteriano.
Se recomiendan inyecciones de dosis altas de inmunoglobulina, en una cantidad de 1-2 mg por kilogramo de peso del paciente por día.
La plasmaféresis se puede prescribir como parte del tratamiento patogénico para controlar la hipercitocinemia.
La base del tratamiento es la esplenectomía y el trasplante de médula ósea de donante.
Prevención
Los expertos actualmente no disponen de información clara sobre los métodos de prevención del síndrome hemofagocítico primario, ya que no se han estudiado completamente las causas del desarrollo de esta patología.
En cuanto al síndrome hemofagocítico secundario, las medidas preventivas pueden incluir las siguientes:
- tratamiento competente y oportuno de infecciones virales y microbianas;
- Tratamiento cualificado de patologías autoinmunes bajo la supervisión de un médico especialista en reumatología.
Pronóstico
El pronóstico del síndrome hemofagocítico se considera extremadamente desfavorable, como lo demuestran las estadísticas: seis fallecimientos en siete pacientes. La supervivencia máxima actual es de dos años.
El síndrome hemofagocítico se considera una enfermedad muy compleja e insidiosa, que hoy en día “compite” sólo con la infección por el virus de la inmunodeficiencia humana, y en términos de frecuencia de consecuencias incluso supera al VIH.
 [ 27 ]
[ 27 ]