Médico experto del artículo.

Nuevos artículos
Encefalomiopatía necrotizante subaguda de Leah
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
La enfermedad se mencionó por primera vez en 1951. Hasta la fecha, se han descrito más de 120 casos. La enfermedad de Leigh (OMIM 256000) es una enfermedad genéticamente heterogénea que puede heredarse nuclearmente (autosómica recesiva o ligada al cromosoma X) o mitocondrialmente (menos frecuente).
 [
[ Causas del síndrome de Leah
La enfermedad se basa en una deficiencia de enzimas que proporcionan producción de energía, principalmente debido a una alteración del metabolismo del ácido pirúvico y un defecto en el transporte de electrones en la cadena respiratoria. Se desarrolla una deficiencia del complejo piruvato deshidrogenasa (subunidad a-E1), piruvato carboxilasa, complejo 1 (NAD-coenzima Q-reductasa) y complejo 4 (citocromo oxidasa) de la cadena respiratoria.
Se ha establecido que los defectos de la piruvato carboxilasa, complejo 1 (NAD-coenzima Q-reductasa) y complejo 4 (citocromo oxidasa) de la cadena respiratoria se heredan de manera autosómica recesiva, los defectos del complejo piruvato deshidrogenasa (subunidad a-E1) se heredan de manera recesiva ligada al cromosoma X. En caso de mutaciones puntuales del mtADN, que afectan a la sexta subunidad de la ATPasa, la herencia mitocondrial es típica. Lo más frecuente es que se produzca una mutación miscens, asociada al reemplazo de timina por guanina o citosina en la posición 8993 del mtADN. Menos común es una mutación en la posición 9176 del mtADN. Debido a que la mutación T8993G es el principal defecto del síndrome NARP, se han descrito familias con estas dos enfermedades. En niños también se ha descrito una mutación en el mtADN en la posición 8344, que se produce en el síndrome MERRF.
Se supone que la acumulación de ADNmt mutante en la mayoría de las mitocondrias provoca un síndrome de Leigh grave. En la génesis mitocondrial de esta afección, el ADNmt mutante se encuentra en el 90 % de las mitocondrias. La patogénesis se asocia con una alteración de la producción de energía en las células y el desarrollo de acidosis láctica.
Síntomas del síndrome de Leah
Los primeros signos de la enfermedad debutan a una edad temprana (1-3 años). Sin embargo, hay casos conocidos de manifestación de la enfermedad a las 2 semanas y a los 6-7 años de edad. Al principio, se desarrollan trastornos inespecíficos: retraso en el desarrollo psicomotor, disminución del apetito, episodios de vómitos, déficit de peso corporal. Posteriormente, los síntomas neurológicos aumentan: hipotonía o distonía muscular con transición a hipertonía, ataques de mioclonías o convulsiones tónico-clónicas, temblor de las extremidades, coreoatetosis, trastorno de coordinación, disminución de los reflejos tendinosos, letargo, somnolencia. La neurodegeneración cerebral es progresiva. Los síntomas de insuficiencia piramidal y extrapiramidal aumentan, el acto de tragar se ve afectado. Se observan a menudo cambios en el órgano de la visión como ptosis, oftalmoplejía, atrofia de los nervios ópticos, con menos frecuencia degeneración pigmentaria de la retina. A veces se desarrolla miocardiopatía hipertrófica, aparecen episodios de taquipnea.
En raras ocasiones, la enfermedad se presenta como una encefalopatía aguda. Es más común un curso crónico o subagudo, que resulta en la muerte varios años después de su inicio. Con una evolución rápida (varias semanas), la muerte se produce por parálisis del centro respiratorio.
Diagnostico del síndrome de Leah
Un análisis bioquímico de sangre revela acidosis láctica debido a la acumulación de ácidos láctico y pirúvico en la sangre y el líquido cefalorraquídeo, así como a un aumento del contenido de alanina en sangre. El nivel de cuerpos cetónicos también puede estar elevado. Se detecta un aumento de la excreción de ácidos orgánicos en la orina: láctico, fumárico, etc. El nivel de carnitina en la sangre y los tejidos suele disminuir.
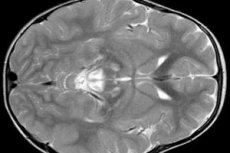
Los resultados del EEG revelan signos focales de actividad epiléptica. Los datos de la RMN revelan agrandamiento de los ventrículos cerebrales, daño cerebral bilateral y calcificación de los ganglios basales (núcleo caudado, putamen, sustancia negra, globo pálido). También se puede detectar atrofia de los hemisferios cerebrales y de la masa encefálica.
El examen morfológico revela cambios macroscópicos en la masa cerebral: focos simétricos de necrosis, desmielinización y degeneración esponjosa del cerebro, principalmente en las secciones medias, la protuberancia anular, los ganglios basales, el tálamo y el nervio óptico. El cuadro histológico incluye degeneración quística del tejido cerebral, gliosis astrocítica, muerte neuronal y un aumento del número de mitocondrias en las células. En los músculos esqueléticos, se observa acumulación de inclusiones lipídicas, disminución de la reacción histoquímica a los complejos 1 y 4 de la cadena respiratoria, acumulación subsarcolemal de mitocondrias y mitocondrias anormales con desorganización de las crestas. El fenómeno de la RRF a menudo no se detecta.
Cómo examinar?
¿Qué pruebas son necesarias?
Использованная литература