Médico experto del artículo.

Nuevos artículos
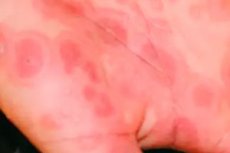
Síndrome de Stevens-Johnson
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
 [
[ Causas Síndrome de Stevens-Johnson
Las posibles causas de la enfermedad suelen ser fármacos (antibióticos, sulfonamidas, analgésicos, barbitúricos, etc.). La enfermedad también puede presentarse debido a otros alérgenos. Actualmente, la mayoría de los dermatólogos creen que la base del eritema exudativo multiforme, así como del síndrome de Stevens-Johnson y del síndrome de Lyell, son reacciones tóxico-alérgicas. Clínica y patogénicamente, la diferencia entre ellos no es cualitativa, sino cuantitativa. En este caso, la reacción antígeno-anticuerpo se dirige a los queratinocitos, con la formación de inmunocomplejos circulantes en el suero sanguíneo y el depósito de IgM y el componente C3 del complemento a lo largo de la membrana basal de la epidermis y la parte superior de la dermis.
Síntomas Síndrome de Stevens-Johnson
La enfermedad se caracteriza por un inicio rápido con síntomas generales pronunciados (fiebre alta, artralgia, mialgia). Tras unas horas o 2-3 días, aparecen lesiones en la piel y las mucosas.
En la piel del tronco, las extremidades superiores e inferiores, la cara y los genitales aparecen manchas eritematoedematosas redondeadas y diseminadas de color carmesí, con una zona periférica y un centro cianótico hundido, de entre 1 y 3,5 cm de diámetro. Estos elementos de la erupción se asemejan a las erupciones de un eritema exudativo multiforme. En el centro de muchas de ellas se forman ampollas flácidas de paredes delgadas con contenido seroso o hemorrágico. Al fusionarse, las ampollas alcanzan enormes dimensiones. Al abrirse, dejan erosiones dolorosas, jugosas y de color rojo brillante, en cuyos bordes se encuentran fragmentos de las cubiertas de las ampollas («collar epidérmico»). Al tocarlas ligeramente, la epidermis se desliza (síntoma de Nikolsky positivo). Con el tiempo, la superficie de la erosión se cubre con costras de color marrón amarillento o hemorrágicas.
En la mucosa oral y ocular se observa hiperemia, edema y aparición de ampollas flácidas, tras cuya apertura se forman erosiones grandes y dolorosas. El borde rojo de los labios está marcadamente edematoso e hiperémico, presenta grietas sangrantes y está cubierto de costras. Con frecuencia se presentan desde blefaroconjuntivitis hasta panoftalmia, con daño en la mucosa uretral, vesical y del tracto respiratorio superior. Los síntomas generales graves (fiebre, malestar general, cefalea, etc.) persisten durante 2-3 semanas.
Diagnostico Síndrome de Stevens-Johnson
En la sangre se observan leucocitosis, eosinopenia, aumento de la VSG, aumento de los niveles de bilirrubina, urea, aminotransferasas, cambios en la actividad fibrinolítica plasmática debido a la activación del sistema proteolítico y una disminución de la cantidad total de proteínas debido a las albúminas.
¿Qué es necesario examinar?
Cómo examinar?
¿Qué pruebas son necesarias?
Diagnóstico diferencial
¿A quién contactar?
Tratamiento Síndrome de Stevens-Johnson
Los corticosteroides se prescriben a una dosis de 1 mg/kg de peso corporal del paciente (en términos de prednisolona) hasta lograr un efecto clínico pronunciado, seguido de una reducción gradual de la dosis hasta su suspensión completa en un plazo de 3 a 4 semanas. Si la administración oral es imposible, se prescriben corticosteroides por vía parenteral. También se toman medidas para inactivar y eliminar antígenos y complejos inmunes circulantes del organismo mediante enterosorbentes, hemosorción y plasmaféresis. Para combatir el síndrome de intoxicación endógena, se administran de 2 a 3 litros de líquido al día por vía parenteral (solución salina, hemodesis, solución de Ringer, etc.), así como albúmina y plasma. También se prescriben calcio, potasio, antihistamínicos y, en caso de riesgo de infección, antibióticos de amplio espectro. Por vía externa se utilizan cremas con corticosteroides (Lorinden C, Dermovate, Advantan, Triderm, Celestoderm con garomicina, etc.), soluciones acuosas al 2% de azul de metileno y violeta de genciana.