Médico experto del artículo.

Nuevos artículos
Síndrome de Usher
Último revisado: 23.04.2024

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
El síndrome de Usher es una enfermedad hereditaria que se manifiesta en forma de sordera completa desde el nacimiento, así como ceguera progresiva con la edad. La pérdida de visión está asociada con la retinitis pigmentaria: este es el proceso de degeneración pigmentaria de la retina del ojo. Muchas personas con síndrome de Usher también tienen serios problemas de equilibrio.
Epidemiología
Debido a la investigación, fue fácil establecer que alrededor del 8% de los niños sordos-mudos examinados estaban enfermos con el síndrome de Usher (las pruebas se realizaron en instituciones especiales para personas sordomudas). Se observó retinitis pigmentada en el 6-10% de los pacientes con sordera congénita, que a su vez se observa en aproximadamente el 30% de las personas con enfermedad pigmentaria de la retina.
Se cree que esta enfermedad se manifiesta en aproximadamente 3-10 personas de 100 mil en todo el mundo. Se puede observar tanto en mujeres como en hombres. Este síndrome afecta a alrededor del 5-6% de la población mundial. Alrededor del 10% de todos los casos de sordera profunda pediátrica ocurren debido al síndrome de Usher I, y también tipo II.
En los Estados Unidos, los tipos 1 y 2 son los tipos más comunes. Juntos, representan aproximadamente del 90 al 95 por ciento de todos los casos de síndrome de Usher en niños.
Causas de síndrome de Usher
El síndrome Usher I, II y también los tipos III, tienen una causa autosómica recesiva, pero el tipo IV se considera una violación del cromosoma X. Las causas de este síndrome de ceguera, así como la sordera, no se han estudiado lo suficiente. Se supone que las personas con esta enfermedad son hipersensibles a los componentes que pueden dañar la estructura del ADN. Con esta enfermedad, los trastornos del sistema inmune también pueden estar asociados, pero en este caso no hay una imagen precisa de dicho proceso.
En 1989, los pacientes con enfermedad tipo II fueron diagnosticados por primera vez con anomalías cromosómicas, lo que eventualmente conduciría a una forma de aislar los genes que desencadenan el desarrollo del síndrome. Además, también será posible identificar estos genes de sus portadores y desarrollar pruebas genéticas prenatales especiales.
 [8]
[8]
Factores de riesgo
La herencia del síndrome ocurre en el caso en que ambos padres están enfermos, es decir, la herencia es de tipo recesivo. Un niño también puede heredar una enfermedad en caso de que sus padres sean portadores del gen. Si ambos futuros padres tienen este gen, la probabilidad de tener un bebé con este síndrome es de 1 a 4. Se considera que una persona que tiene un solo gen del síndrome es portadora, pero él mismo no tiene los síntomas del trastorno. Hoy en día aún no es posible determinar si una persona tiene un gen para esta enfermedad.
Si el hijo nace de padres, uno de los cuales no tiene dicho gen, entonces la probabilidad de que él herede el síndrome es muy baja, pero será un portador inequívoco.
Síntomas de síndrome de Usher
Los síntomas del síndrome de Usher son la pérdida de la audición y además de esta acumulación patológica de células pigmentadas en las estructuras oculares. Además, el paciente desarrolla degeneración de la retina del ojo, debido a lo que comienza el deterioro de la visión con la consiguiente pérdida de la misma en el caso más severo.
La pérdida auditiva neurosensorial es leve o completa y generalmente no progresa desde el nacimiento. Pero la retinitis pigmentosa puede comenzar a desarrollarse en la infancia o más tarde. Los resultados de la encuesta mostraron que la agudeza de la visión central puede persistir durante muchos años, incluso cuando la visión periférica se deteriora (esta afección se denomina "visión de túnel").
Estas son las principales manifestaciones de la enfermedad, que a veces pueden complementarse con otros trastornos, como la psicosis y otros trastornos mentales, problemas con el oído interno y / o las cataratas.
Formas
Durante la investigación, se identificaron 3 tipos de esta enfermedad y también 4 formas, bastante raras.
Mi tipo de enfermedad se caracteriza por una sordera completa congénita, así como un trastorno del equilibrio. A menudo, estos niños comienzan a caminar solo a la edad de 1.5 años. El deterioro de la visión generalmente comienza con 10 años, y el desarrollo final del estado de ceguera nocturna comienza con 20 años. En los niños con este tipo de enfermedad, puede desarrollarse un deterioro progresivo de la visión periférica.
Con la enfermedad de tipo II, se observa sordera moderada o congénita. A menudo, en este caso, el deterioro con sordera parcial ya no se produce. La retinitis pigmentaria comienza a desarrollarse hacia el final del período de la adolescencia o después de los 20 años. El desarrollo de la ceguera nocturna generalmente comienza en el año 29-31. Las alteraciones en la agudeza visual en el caso de la patología tipo II básicamente progresan un poco más lentamente que en el tipo I.
Tipo III enfermedades caracterizadas por la pérdida progresiva de la audición, por lo general comienza en la pubertad, así como la aparición gradual del mismo período (un poco más tarde de sordera) retinitis pigmentosa que puede convertirse en un factor en el desarrollo de ceguera progresiva.
Las manifestaciones del cuarto tipo de patología ocurren principalmente en los hombres. En este caso, también hay trastornos progresivos y pérdida de audición y visión. Esta forma es muy rara y generalmente tiene una naturaleza de cromosoma X.
Diagnostico de síndrome de Usher
El diagnóstico del síndrome de Usher se hace sobre la base de la combinación del paciente de sordera súbita con pérdida progresiva de la visión.
Análisis
Para detectar una mutación, se puede asignar una prueba genética especial.
Se encontraron 11 loci genéticos que pueden causar el desarrollo del síndrome de Usher e identificaron nueve genes que son exactamente la causa del trastorno:
- Tipo 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Tipo 2: ush2a, VLGR1, WHRN.
- Síndrome de Ushira tipo 3: USH3A.
Científicos de NIDCD junto con colegas de universidades en Nueva York e Israel identificaron una mutación llamada R245X del gen Pcdh15, que es un gran porcentaje del 1 tipo de síndrome de Usher entre la población judía.
Para obtener información sobre los laboratorios que realizan ensayos clínicos, visite https://www.genetests.org y busque en el catálogo de investigación de laboratorio escribiendo el término "síndrome de Usher".
Para obtener información sobre los ensayos clínicos actuales que incluyen pruebas genéticas síndrome de Usher, puede visitar el sitio web e introduzca la búsqueda https://www.clinicaltrials.gov "síndrome de Usher" o "pruebas genéticas síndrome de Usher."
[25], [26], [27], [28], [29], [30]
Diagnóstico instrumental
Hay varios métodos de diagnóstico instrumental:
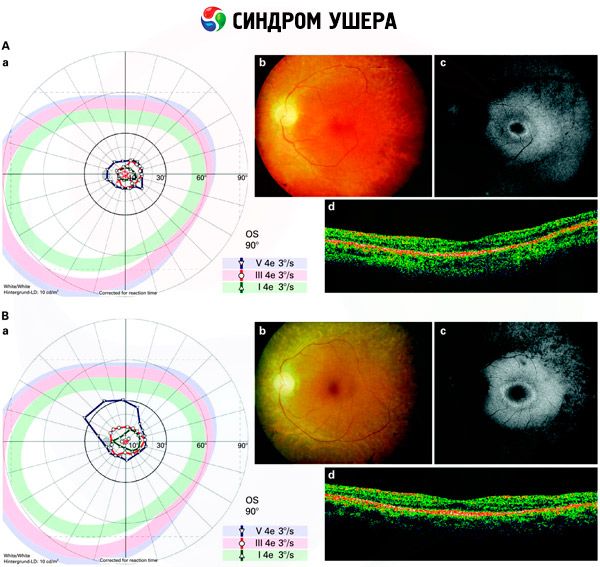
- Examen del fondo de ojo para identificar la presencia de retinas en la retina, así como el estrechamiento de los vasos retinianos;
- Electroretinograma, que le permite identificar las anormalidades degenerativas iniciales en la retina del ojo. Muestra la extinción de las trayectorias electro-radiográficas;
- El electronistagmograma (ENG) mide los movimientos oculares involuntarios, lo que podría indicar la presencia de desequilibrio
- Audiometría, que determina la presencia de sordera y el grado de su gravedad.
Diagnóstico diferencial
El síndrome de Usher debe diferenciarse con algunas anomalías similares.
El síndrome de Hallgren, en el que se observa pérdida de audición congénita, así como la pérdida progresiva de la visión (también hay cataratas y nistagmo). Entre los síntomas adicionales de la enfermedad: ataxia, trastornos psicomotores, psicosis y retraso mental.
Síndrome de Alstrom, que es una enfermedad hereditaria en la que se produce la degeneración de la retina, como resultado de la cual se pierde la visión central. Este síndrome está asociado con el problema de la obesidad infantil. En este caso, la diabetes mellitus y la pérdida de audición comienzan a desarrollarse después de 10 años.
La rubéola en una mujer embarazada en el primer trimestre puede causar una variedad de anomalías en el desarrollo del niño. Entre las consecuencias de esta anomalía está la pérdida de audición, así como (o) problemas con la vista, y además de esto, varios defectos de desarrollo.
¿A quién contactar?
Tratamiento de síndrome de Usher
Para curar el síndrome de Usher ahora es imposible. Por lo tanto, la terapia en este caso es principalmente para retrasar el proceso de caída de la visión y también para compensar la pérdida de la audición. Los posibles tratamientos incluyen:
- El uso del grupo de vitamina A (algunos oftalmólogos creen que las dosis altas de palmitato de vitamina A pueden ralentizar, pero no detener, la progresión de la retinitis pigmentaria);
- Implantación de dispositivos electrónicos especiales en las aurículas del paciente (aparato auditivo, implantes cocleares).
Los oftalmólogos recomiendan que la mayoría de los pacientes adultos con formas avanzadas de pigmentación de la retinitis tomen diariamente 15,000 UI (unidades internacionales) de vitamina A en forma de palmitato bajo supervisión. Dado que las personas con síndrome de Usher tipo 1 no participaron en el estudio, no se recomiendan altas dosis de vitamina A para este grupo de pacientes. Las personas que están considerando tomar vitamina A deben discutir esta opción con su médico. Otras recomendaciones con respecto a esta opción de tratamiento incluyen:
- Cambie su dieta con la inclusión de alimentos ricos en vitamina A.
- Las mujeres que planifican el embarazo deben dejar de tomar altas dosis de vitamina A tres meses antes de la concepción planificada debido al mayor riesgo de defectos de nacimiento.
- Las mujeres que están embarazadas deben dejar de tomar altas dosis de vitamina A debido a un mayor riesgo de defectos de nacimiento.
También es importante adaptar ese niño a la vida social. Esto requiere la ayuda de educadores, defectólogos y psicólogos. En el caso en que el paciente haya comenzado una caída progresiva en la visión, se le debe enseñar a usar el lenguaje de señas.
Pronóstico
El síndrome de Usher tiene un pronóstico desfavorable. El campo de visión y su gravedad comienzan a deteriorarse en el período de 20-30 años en la mayoría de los pacientes con esta enfermedad de cualquier tipo. En algunos casos, se trata de una pérdida de visión bilateral completa. La sordera, que siempre se observa y la mudez, se desarrolla muy rápidamente a una pérdida bilateral completa de la audición.