Médico experto del artículo.

Nuevos artículos
Síndrome de Angelmann en niños y adultos
Último revisado: 23.04.2024

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.

Hay una serie de enfermedades en las que expresiones como "cuídate y no te enfermes" suenan, al menos, ridículas. Esta patología, en la cual algunas anormalidades mentales y físicas están incrustadas en el cuerpo de un niño incluso antes del nacimiento, pero los padres no tienen ninguna culpa. Tales enfermedades son causadas por mutaciones o trastornos en los conjuntos de cromosomas y se llaman cromosómicas o genéticas. Síndrome de Angelman, síndrome de Down, Patau, Edwards, Turner, Prader-Willi son solo parte de las enfermedades genéticas de una lista bastante decente.
Síndrome de una persona feliz
Esta vez hablaremos de una patología que lleva el nombre del pediatra de Inglaterra, Harry Angelmann, que planteó por primera vez el problema de este problema en el año 1 965, habiéndose enfrentado el día anterior en su práctica con tres niños inusuales, unidos por síntomas peculiares comunes. El médico nombró a estos niños niños títeres y escribió sobre ellos un artículo que originalmente se llamaba "Niños títeres". El artículo en sí y su nombre fueron escritos bajo la impresión de una imagen vista en uno de los museos en Verona. La imagen mostraba a un niño riendo, y se llamaba "Niño-marioneta". La asociación del niño representado en la imagen con los tres niños que Angelman una vez encontró en su práctica, y animó al pediatra a unir a los niños en un grupo debido a su enfermedad existente.
El hecho de que los niños notados en el artículo no fueron notados por otros doctores no es sorprendente. Después de todo, a primera vista parecía que tenían enfermedades completamente diferentes, por lo que el cuadro clínico general de la enfermedad difería en 3 casos diferentes. Una "nueva" patología cromosómica podría ser de interés para otros científicos, pero en ese momento la genética aún no estaba lo suficientemente desarrollada como para confirmar la hipótesis de un médico inglés. Por lo tanto, el artículo después de un cierto interés en él durante mucho tiempo fue abandonado al regimiento distante.
La siguiente mención del síndrome de Angelman, por lo que ahora se llamaba el artículo del pediatra de Inglaterra, G. Anglemann, data de principios de los años 80 del siglo XX. Y solo en 1987 fue posible encontrar la razón por la cual una pequeña parte de los niños nacen con tales desviaciones, que por el lado parecen estar constantemente sonrientes y felices. De hecho, esto no es así, y la sonrisa es solo una mueca, detrás de la cual yace el infeliz alma humana y el dolor de los padres.
Epidemiología
La mutación cromosómica en un niño, según las estadísticas, puede desarrollarse tanto en el contexto de tales mutaciones en los padres, y en la ausencia de tal. No hay un claro carácter hereditario en el síndrome de Angelmann (CA), pero la probabilidad de desarrollar patología en padres con mutaciones cromosómicas es bastante alta.
También es interesante que si una familia ya tiene un hijo con una SA, existe un uno por ciento de posibilidades de tener un segundo hijo del mismo tipo, incluso si los padres están sanos.
Todavía no hay estadísticas exactas sobre el número de pacientes con síndrome de Anghelman. Tal vez la falla es una variedad de síntomas que pueden ocurrir en una determinada composición o que durante mucho tiempo no surgen en absoluto. Se supone que la prevalencia de la enfermedad es: 1 niño por cada 20,000 recién nacidos. Pero esta figura es muy aproximada.
Causas de síndrome de angelman
El síndrome de Angelmann es el nombre médico de la patología cromosómica, pero de ninguna manera es el único. En las personas, esta enfermedad también se conoce como el síndrome de los niños títeres, y el síndrome de un títere feliz, y el síndrome de Petrushka, y el síndrome de una muñeca que se ríe. Sí, qué tipo de nombres no se le ocurren a las personas (a veces incluso insultando a los pacientes mismos y a sus padres), pero la enfermedad es una enfermedad, sin importar cuán divertida parezca exteriormente y sean cuales sean los motivos que puedan ser causados.
Y las razones para el desarrollo del síndrome de Angelman, así como de muchas otras patologías genéticas, en todos los casos son violaciones en la estructura de uno de los cromosomas o en el conjunto de los cromosomas. Pero solo en nuestro caso, todo el problema radica en los 15 cromosomas transmitidos por la madre. Es decir. El cromosoma paterno en este caso no tiene desviaciones, pero la mujer sufre ciertas mutaciones.
De acuerdo con el tipo de anormalidad cromosómica, el síndrome de Angelmann se refiere a las mutaciones cromosómicas. Tales mutaciones son:
- La deleción (ausencia de la región cromosómica que contiene un conjunto específico de genes, y si no hay uno de los genes se trata microdeleciones), que es el resultado de dos discontinuidades y una reunificación cuando se pierde parte del cromosoma originales.
- Duplicación (la presencia de un sitio extra en el cromosoma, que es una copia del ya disponible), que en la mayoría de los casos conduce a la muerte de una persona, con menos frecuencia, a la infertilidad.
- Inversión (inversión de un secciones de cromosomas a 180 grados, es decir, en la dirección opuesta, y entonces los genes en el mismo están dispuestos en orden inverso) cuando el cromosoma roto extremos conectados en un orden diferente de la original.
- Inserción (si parte del material genético en el cromosoma no está en su lugar),
- translocación (si una parte del cromosoma se une a otro cromosoma, tal mutación puede ser mutua sin pérdida de sitios).
Al obtener un cromosoma mutado de una madre desprevenida, el bebé está condenado a nacer con desviaciones por adelantado. La causa más común del desarrollo del síndrome de Anghelman sigue siendo la eliminación del cromosoma 15 materno, cuando no hay un área pequeña en el mismo. Las mutaciones menos comunes en el síndrome de la "muñeca que ríe" son:
- translocación,
- Diosomia de un solo padre (si el niño recibió un par de cromosomas del padre, el cromosoma madre está ausente)
- mutación de genes en el ADN, que son tanto el material de construcción principal (material genético) como las instrucciones para su uso correcto (en particular, la mutación del gen ube3a en el cromosoma materno).
La presencia de una de esas mutaciones en los padres es un factor de riesgo para el síndrome de Anghelman en niños. Pero no solo las mutaciones cromosómicas, sino también las mutaciones genómicas (que están asociadas con un cambio cuantitativo en los conjuntos de cromosomas y se encuentran con más frecuencia que los cromosomas) pueden desencadenar el desarrollo de la enfermedad en el niño. A las mutaciones genómicas comunes se les puede atribuir la trisomía de los cromosomas (si una persona tiene un conjunto de cromosomas tiene más de 46 cromosomas).
Para la patología del bebé no necesariamente tiene que tener padres anormalidades cromosómicas. Y, sin embargo, existe un cierto porcentaje de pacientes cuya enfermedad es hereditaria.

Patogenesia
Vamos a cavar un poco en biología, más precisamente en genética. La información genética de cada cuerpo humano está contenida en 23 pares de cromosomas. Un cromosoma del par se transmite al niño del padre y el otro a la madre. Todos los pares de cromosomas difieren en forma y tamaño y llevan consigo cierta información. Entonces, 23 pares de cromosomas (cromosomas X e Y) son responsables de la formación de las características sexuales del niño (XX - una niña, XY-boy, mientras que un niño puede obtener un cromosoma Y solo de su padre).
Idealmente, un niño recibe de sus padres 46 cromosomas, que forman sus atributos genéticos, predeterminándolo como individuo. Un mayor número de cromosomas se llama trisomía y se considera una desviación de la norma. Por ejemplo, la presencia de 47 cromosomas en el conjunto de cromosomas (cariotipo, que determina las especies y las características individuales) causa la aparición del síndrome de Down.
Si los cromosomas están teñidos con un tinte especial, entonces en el microscopio se pueden ver bandas de diferentes tonos a lo largo de cada uno de ellos. Dentro de cada banda hay una gran cantidad de genes. Todas estas bandas son numeradas por científicos y tienen una ubicación fija. La ausencia de una de las bandas se considera una desviación de la norma. Con el síndrome de Angelmann, muy a menudo se puede observar la ausencia de segmentos del cromosoma materno en el intervalo q11-q13 ubicado en el brazo largo, el número de bases de ADN en el que solo hay unos 4 millones.
El componente principal del cromosoma es una molécula de ADN increíblemente larga que contiene miles de genes y decenas y cientos de millones de bases nitrogenadas. Por lo tanto, los 15 cromosomas responsables del desarrollo del síndrome de Angelman y varios otros, contienen 1200 genes y aproximadamente 100 millones de bases. Cualquier violación en la estructura de la molécula de ADN necesariamente afectará la apariencia y el desarrollo del feto.
La información genética contenida en los genes se convierte en una proteína o ARN. Este proceso se llama expresión génica. Por lo tanto, la información genética recibida de los padres recibe tanto forma como contenido, encarnado en su único heredero del sexo femenino o masculino.
Hay una serie de patologías con un tipo de herencia no clásica, incluido el síndrome de Angelmann, en el que los genes obtenidos de los padres en los cromosomas pares tienen una huella peculiar de los padres y se manifiestan de diferentes maneras.
Así, el síndrome de Angelman es un ejemplo típico de la impronta genómica, mediante el cual la expresión de genes en el cuerpo del niño es dependiendo directamente sobre de quien alelos parentales derivado (diferentes formas de la misma gen obtenido a partir del padre y la madre están situadas en las porciones idénticas de los cromosomas emparejados) . Es decir. A la aparición del síndrome solo conducen anomalías en el cromosoma materno, mientras que las mutaciones y alteraciones en la estructura del cromosoma paterno causan patologías completamente diferentes.
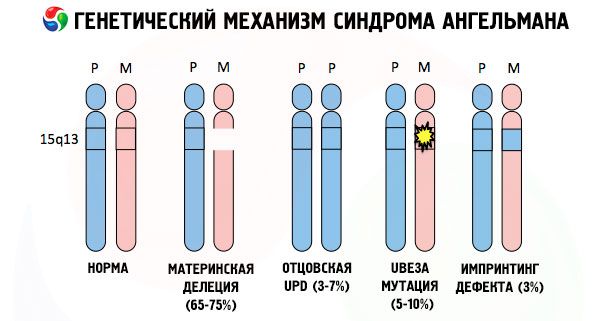
En esta enfermedad hay una falta de genes específicos en el cromosoma materno o la pérdida / disminución en la actividad de genes individuales (en la mayoría de genes casos UBE3A implicada en el metabolismo de ubiquitina - la degradación de proteínas otras proteínas reguladoras). Como resultado, el niño es diagnosticado con anomalías de desarrollo mental y deformidades físicas.
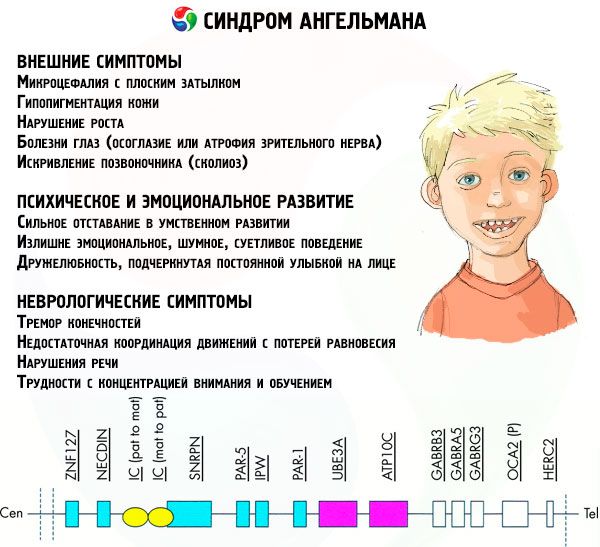
Síntomas de síndrome de angelman
La sintomatología del síndrome de Angelman afecta varios aspectos de la vida y el desarrollo de un niño: físico, neurológico y psíquico. En base a esto, podemos distinguir 3 grupos de síntomas que indican el desarrollo de esta patología.
- Síntomas externos o físicos:
- una cabeza desproporcionadamente pequeña en comparación con el tronco y las extremidades que son de tamaño normal,
- boca muy ancha,
- en la cara casi siempre hay una sonrisa (con la boca abierta),
- dientes raros,
- labio superior estrecho,
- a menudo sacando una lengua ancha,
- prominente mandíbula inferior,
- barbilla afilada,
- piel muy clara, a menudo también pelo (albinismo, asociado con el hecho de que el cuerpo no produce el pigmento melanina),
- manchas oscuras en la piel clara (hipopigmentación debido a la producción insuficiente de melanina)
- síntomas físicos o externos: enfermedades oculares como estrabismo o atrofia del nervio óptico,
- curvatura de la columna vertebral (escoliosis),
- piernas rígidas (cuando camina un hombre no dobla las rodillas debido a la pequeña movilidad de las articulaciones, de ahí la comparación con la marcha de marionetas).
- Síntomas asociados con el desarrollo mental y emocional:
- un fuerte retraso en el desarrollo mental,
- comportamiento innecesariamente emocional, ruidoso, quisquilloso,
- aplausos frecuentes
- la amabilidad expresada, subrayada por una sonrisa constante en la cara,
- risa frecuente y sin causa.
- Síntomas neurológicos:
- temblor de extremidades,
- coordinación insuficiente de movimientos con pérdida de equilibrio,
- disminución del tono muscular,
- una variedad de trastornos del sueño,
- frecuentes convulsiones histéricas en la infancia,
- discapacidad del habla (el niño comienza a hablar tarde, tiene poca capacidad de comunicación y dificultad para hablar)
- hiperactividad en un contexto de aumento de la excitabilidad,
- dificultades con la concentración y el entrenamiento.
Pero esta es una imagen generalizada de la enfermedad. De hecho, el cuadro clínico del síndrome de Anghelman depende en gran medida del estadio de desarrollo de la enfermedad y del tipo de mutación cromosómica que causó la patología. Y esto significa que en diferentes pacientes la sintomatología de la enfermedad puede diferir significativamente, lo que durante mucho tiempo no permitió aislar la patología entre otros con un cuadro clínico similar.
Entre el número total de síntomas se pueden identificar los que son característicos de todos los pacientes sin excepción:
- desviaciones severas en el desarrollo mental,
- comportamiento inadecuado (risa sin causa, aumento de la excitabilidad, falta de concentración de la atención, un estado de euforia),
- subdesarrollo de las habilidades motoras,
- mala coordinación de movimientos, ataxia de caminar (ritmo desigual, balanceo de un lado a otro, etc.), temblor de extremidades.
- violación del desarrollo del habla con predominio de medios de comunicación no verbales.
Entre los síntomas que ocurren en la gran mayoría de los pacientes, podemos distinguirlos:
- cabeza y tronco desproporcionados, causados por un retraso en el desarrollo físico,
- en muchos pacientes, la forma del cráneo es tal que el tamaño del cerebro sigue siendo más pequeño que en personas sanas (microcefalia),
- ataques epilépticos en la edad de hasta 3 años con una disminución progresiva de la fuerza y la frecuencia en los ancianos,
- distorsión de los índices de EEG (oscilaciones y alta amplitud de las ondas de baja frecuencia).
Estos síntomas ocurren con bastante frecuencia, sin embargo, en el 20% de los pacientes con síndrome de Angelmann están ausentes.
Aún más raramente se pueden diagnosticar tales manifestaciones de la enfermedad como:
- estrabismo pronunciado o leve,
- un control débil sobre el movimiento de la lengua, como resultado de lo cual los pacientes a menudo sacan la lengua sin razón,
- dificultades para tragar y succionar, especialmente en niños más pequeños,
- violación de la pigmentación de la piel y los ojos,
- levantado o doblado durante caminar manos,
- giperperflexia,
- trastornos del sueño, especialmente en la infancia,
- salivación frecuente,
- Sed incontenible
- movimientos de masticación excesivamente activos,
- hipersensibilidad al calor,
- cabeza plana,
- mandíbula inferior avanzada,
- palmas suaves.
Un gran porcentaje de pacientes tienen problemas para orinar, lo que controlan mal, la violación de las habilidades motoras finas, lo que crea dificultades en el autoservicio y el entrenamiento, el sobrepeso. Prácticamente en todos los pacientes, la pubertad comienza más tarde que en los pares sanos.
Los niños con síndrome de angelman son buenos para comprender y comprender el habla oral, pero no quieren participar en la conversación, lo que limita su discurso a docenas de palabras necesarias en la vida cotidiana. Pero en la edad adulta, estos pacientes parecen más jóvenes que sus pares sin patologías genéticas.
Muchos de los síntomas del síndrome de Angelmann son inconstantes, por lo que el cuadro clínico de la enfermedad cambia con la edad. Las convulsiones y las convulsiones epilépticas se vuelven más raras o desaparecen, el paciente se vuelve menos agitado, se instala el sueño.
Complicaciones y consecuencias
El síndrome de Angelman es una patología cromosómica grave, prácticamente incurable hasta la fecha, que priva a los pacientes de la oportunidad de vivir una vida normal. Cuál será la vida del bebé con la SA, depende en gran medida del tipo de anormalidad cromosómica.
La duplicación de la región cromosómica en la mayoría de los casos es incompatible con la vida. E incluso si tales pacientes no mueren en la infancia y alcanzan la pubertad, no tienen posibilidad de tener hijos.
La eliminación o ausencia de una parte de los genes que ocurre en el síndrome de Anghelman con mayor frecuencia es un obstáculo para que el niño aprenda a caminar y hablar. En tales niños, el retraso mental se presenta en una forma más grave, a menudo se producen ataques de epilepsia y su intensidad es mucho más fuerte que en pacientes con otras anomalías cromosómicas.
Si solo hay una mutación de un gen, con la atención y el enfoque adecuados, al niño se le pueden enseñar los conceptos básicos del autoservicio, la comunicación y la comunicación en el equipo, aunque aún se retrasará en el desarrollo de sus compañeros.
Para los niños con síndrome de Angelmann, benévolos por naturaleza, lo principal es el amor y la atención de los padres. Solo en este caso el entrenamiento del niño dará sus frutos, incluso los pequeños. Por supuesto, los pacientes en la escuela regular no podrán estudiar con la SA. Necesitan clases especiales, donde primero se enseñe a los niños a concentrar su atención, y luego darán gradualmente los conocimientos básicos sobre el conocimiento de la escuela.
Diagnostico de síndrome de angelman
El síndrome de Angelman es una patología congénita del desarrollo. Pero debido a algunas circunstancias, el diagnóstico en la infancia y la primera infancia a menudo no es posible. La razón de esto es la inespecificidad y los síntomas leves en bebés y niños pequeños de hasta 3 años. Y la prevalencia de la enfermedad en nuestro país no es tan grande como para que los médicos la hayan aprendido a reconocerla.
El síndrome de Angelman en bebés puede manifestarse en forma de disminución del tono muscular, lo que se manifiesta en forma de problemas de alimentación (debilidad para tragar y beber reflejo), y dificultades de aprendizaje más tarde a caminar (tales niños mucho más tarde empiezan a caminar). Estos síntomas son los primeros signos de una desviación en el desarrollo del bebé, que bien puede estar asociada con una anomalía cromosómica. Confirme esta suposición solo puede análisis genético.
Se presta especial atención a los niños cuyos padres tienen diferentes anomalías genómicas o cromosómicas. Después de todo, la enfermedad no puede manifestarse de ninguna manera, y si la patología se revela a tiempo, al comenzar a trabajar duro con el niño, es posible lograr un éxito mucho mayor en el entrenamiento, disminuyendo la progresión de la enfermedad.
Si los padres tienen diferentes anomalías cromosómicas, el análisis genético se lleva a cabo incluso antes de que nazca el bebé, ya que la CA es una de las patologías que se pueden detectar en un estado embrionario.
La recolección de material para la investigación genética se puede hacer de dos maneras:
- invasivo (con un cierto porcentaje de riesgo, porque se requiere ingresar al útero para tomar una prueba de líquido amniótico),
- no invasivo (análisis de ADN del bebé por la sangre de la madre).
Luego se llevan a cabo las siguientes investigaciones:
- hibridación fluorescente in situ (método FISH): unión de una sonda de ADN marcada con un colorante especial al ADN en estudio, seguida de un examen microscópico.
- análisis de mutaciones en el gen ube3a y genes de impronta,
- análisis de la metilación del ADN con la ayuda de métodos especiales utilizados en genética.
Los análisis genéticos brindan información bastante precisa en el caso de anormalidades cromosómicas, por lo que los futuros padres saben de antemano para qué deberían estar preparados. Sin embargo, hay excepciones. En cierto grupo de pacientes, en presencia de todos los síntomas que indican síntomas, los resultados de los análisis permanecen normales. Es decir. Para revelar una patología es posible observar de cerca al niño desde la primera infancia: cómo se come, cuándo comenzó a caminar y hablar, si las piernas se doblan al caminar, etc.
Además de PEZ método, entre los métodos para la herramienta de diagnóstico síndrome de Angelman puede ser distinguido de tomografía (CT o MRI), ayudando a determinar el estado y el tamaño del cerebro, y el electroencefalograma (EEG), que muestra cómo las partes individuales del cerebro de trabajo.
El diagnóstico final de los médicos generalmente se establece a la edad de 3-7 años, cuando el paciente ya tiene la mayoría de los síntomas y la dinámica del desarrollo de la enfermedad es visible.
¿Qué pruebas son necesarias?
Diagnóstico diferencial
El síndrome de Angelman es una patología genética que en realidad no tiene manifestaciones específicas. La mayoría de los síntomas pueden indicar igualmente CA y otras patologías genéticas.
El diagnóstico diferencial en el síndrome de Anghelman se lleva a cabo con las siguientes patologías:
- Síndrome de Pitt-Hopkins (los pacientes se caracterizan por retraso mental, carácter alegre, sonríen, tienen una boca bastante grande y ancha, se observa microcefalia). Diferencia: ataques de hiperventilación y retraso de la respiración en estado de vigilia.
- Síndrome de Kristiansona (pacientes mentalmente retrasados con una disposición alegre, incapaces de hablar, se caracterizan por microcefalia, ataxia, convulsiones, movimientos involuntarios de los músculos).
- Síndrome de Mowata-Wilson (síntomas: retraso mental, ataques epilépticos, barbilla aguda, boca abierta, expresión de felicidad en la cara, microcefalia). La diferencia es una gran distancia entre los ojos, los ojos están achaflanados hacia el interior, la punta de la nariz es redondeada, la aurícula está vuelta hacia atrás.
- Síndrome de Kabuki (caracterizado por un grado leve y moderado de retraso mental, problemas del habla y del motor, debilidad muscular, ataques epilépticos, microcefalia, intervalos grandes entre prurito, coordinación alterada). Diferencia: cejas en forma de arco, parte lateral invertida del párpado inferior, ojos muy separados, hendiduras oculares largas con pestañas largas y gruesas.
- Síndrome de Rett (diferenciación con CA en mujeres). Síntomas: retraso en el desarrollo del habla, ataques convulsivos, microcefalia. La diferencia: no hay una expresión feliz en la cara, hay ataques de apnea y apraxia, que eventualmente progresan.
- Síndrome de retraso mental autosómico recesivo 38 (síntomas: retardo mental marcado con retraso en el desarrollo de habilidades motrices y del habla, debilidad muscular, problemas con la alimentación en la infancia, impulsividad). La diferencia es el color azul del iris.
- Síndrome de duplicación del gen MESR 2 (diferenciación con SA en hombres). Síntomas: retraso mental severo, debilidad muscular desde la niñez, problemas con el habla o falta de ella, epilepsia. Diferencias: miopatía progresiva, infecciones constantemente recurrentes.
- Síndrome de Clifstra (sintomático: problemas del habla y del pensamiento, debilidad muscular, trastornos del sueño, falta de atención, boca ligeramente abierta, hiperactividad, convulsiones, ataxia, desequilibrio). Diferencias: cara plana, nariz corta y chata, ojos muy abiertos, labio inferior grande invertido, ataques de agresión.
- Síndrome de Smith-Magenis (caracterizado por convulsiones, problemas con el sueño, trastornos del desarrollo intelectual y motor). Diferencias: una cara ancha y plana, frente convexa.
- Síndrome de Kulena-de Vries (retraso mental leve y moderado, debilidad muscular, ataques convulsivos, amabilidad). Diferencias: una cara larga con frente alta, orejas salientes, ojos oblicuos, mayor movilidad de las articulaciones, patologías cardíacas congénitas.
- Síndrome de Philan - McDermid (síntomas: retraso mental, deterioro del habla o falta de eso). Diferencias: manos grandes con músculos desarrollados, debilidad muscular desde el nacimiento, sudoración débil.
El síndrome de Angelman síntomas similares pueden "alarde" y como una deficiencia adenilsuktsinazy tal patología, un síndrome autosómico recesivo de retraso mental 1, el síndrome de la duplicación del cromosoma 2q23.1, los genes haploinsuficiencia FOXG1, STXBP1 o MEF2C y otros.
La tarea del médico es hacer un diagnóstico preciso, diferenciando el síndrome de Angelmann de patologías con síntomas similares y prescribir un tratamiento efectivo que sea relevante para el grado de desarrollo de la enfermedad diagnosticado.
Tratamiento de síndrome de angelman
El síndrome de Angelman se refiere a la categoría de esas patologías, la búsqueda de un tratamiento efectivo del cual la medicina está comprometida hasta el día de hoy. El tratamiento etiológico de la enfermedad se encuentra en la etapa de desarrollo de varios métodos y medios, muchos de los cuales aún no se han probado en humanos. Hasta ahora, los médicos tienen que limitarse a la terapia sintomática de alguna manera para ayudar a aliviar el sufrimiento de los niños y adultos con síndrome de marionetas, que sufre de ataques epilépticos, salivación, hipotensión y trastornos del sueño.
Por lo tanto, reduzca la frecuencia y la fuerza de las convulsiones epilépticas con un medicamento anticonvulsivo seleccionado adecuadamente. Pero toda la dificultad es que las convulsiones en pacientes con EA difieren de las convulsiones epilépticas habituales en que se caracterizan por varios tipos de convulsiones, lo que significa que será posible aliviar la afección mediante la administración de varios fármacos a la vez.
Los anticonvulsivos más populares usados para tratar el síndrome de Anghelman son: ácido valproico, topiramato, lamotrigina, levetiracetam, clonazepam y preparaciones basadas en ellos. Menos comúnmente utilizado medicamentos basados en karmazepina, fenitoína, fenobarbital, etosuximida, porque algunos de ellos podrían desencadenar un efecto paradójico, es reforzar y aumentar la frecuencia de las crisis epilépticas. Esto sucede si el medicamento se usa como parte de la monoterapia.
Para el tratamiento de la salivación, se utilizan dos métodos: medicinal (preparaciones que suprimen la formación de saliva) y operativo, que consiste en la reimplantación de los conductos salivales. Pero en el caso de CA, estos métodos se consideran ineficaces, y la pregunta permanece abierta. Los padres y las personas que cuidan a tales pacientes, debemos prestar especial atención a este momento, ya que los pacientes mismos generalmente no controlan la salivación, y algunos simplemente no pueden cuidarse a sí mismos.
Otro problema es la corta duración del sueño. A menudo, los niños con síndrome de Angelman no duermen más de 5 horas, lo que afecta negativamente el trabajo de todo el organismo. Los niños excitables y activos, los juegos amorosos y la comunicación (incluso si intentan limitarse a formas no verbales), están notablemente cansados por el día. Para tener un buen descanso, el cuerpo necesita un sueño profundo, pero ese es solo el problema.
Parecería que para mejorar el sueño en pacientes excitables deberían existir suficientes fármacos con efecto sedante (fenotiazinas y antipsicóticos atípicos), calmando el sistema nervioso. Pero en el caso de CA, el uso de tales drogas está cargado con la aparición de efectos negativos. Por lo tanto los médicos prefieren fármacos hipnóticos todavía luz, tales como "melatonina" (preparación hormonal natural sobre la base de la hormona del sueño), que dan a los pacientes una hora antes de ir a la cama a 1 tableta, y "difenhidramina". La frecuencia de administración y dosificación la establece el médico, dependiendo de la condición y la edad del paciente.
A veces, los pacientes con síndrome de angelman tienen problemas con la digestión y las heces. Para ajustar una silla es posible con preparaciones laxantes (es mejor que una fitogenia).
Y usted puede acercarse al problema de manera diferente, al igual que los médicos estadounidenses, basados en algunos de los métodos de tratamiento del autismo, porque muchos de los síntomas característicos de la SA, también son característicos del autismo (impulsividad, movimientos involuntarios, las acciones repetitivas, déficits de atención, problemas de comunicación, etc. .). Se observó que la administración de la hormona secretina, que normaliza la digestión y las heces, afecta positivamente la atención de los pacientes y la oxitocina ayuda a mejorar las capacidades cognitivas y la memoria del niño, y a corregir el comportamiento.
Es cierto que algunas hormonas son indispensables aquí, especialmente cuando se trata de niños. El síndrome de Angelman muestra terapia conductual, trabaja con un psicólogo y un terapeuta del habla (enseñando formas de comunicación no verbal y lenguaje de señas). La capacitación de tales niños debe basarse en un programa individual con la participación de maestros especialmente capacitados, un psicólogo y padres. Desafortunadamente, esto no es posible en todas partes, y las familias se quedan solas con su problema.
Dado que muchos pacientes pequeños con AC sufren de un bajo tono muscular y problemas en las articulaciones, se presta mucha atención al tratamiento fisioterapéutico. Muy a menudo, los médicos recurren al uso de aplicaciones de parafina, electropreasis, magnetoterapia.
El masaje tonificante activo y los ejercicios especiales de fisioterapia ayudarán al niño enfermo después de un tiempo de pie y caminando confiadamente. Especialmente útil a este respecto aquagymnastics, que se recomienda en CA en agua fría. Aumenta el tono de los músculos y le enseña al bebé a poseer su propio cuerpo, coordinar movimientos.
Tratamiento anticonvulsivo
El síntoma más peligroso en el síndrome de Anghelman son las convulsiones, similares a las convulsiones epilépticas. Este síntoma se observa en el 80% de los pacientes, lo que significa que a todos se les debe prescribir un tratamiento anticonvulsivo eficaz.
El tratamiento de las crisis epilépticas se lleva a cabo con la ayuda de vitaminas y anticonvulsivos. Cuando el síndrome de Angelman, acompañado de síndrome convulsivo, será útil vitaminas del grupo B, así como las vitaminas C, D y E. Sin embargo, la terapia con vitamina nombrar a su propio en este caso es muy peligroso, ya que la ingesta incontrolada de vitaminas puede reducir la eficacia de los fármacos anti-epilépticos y provocar nuevas y más severas y prolongadas convulsiones.
La elección de los fármacos anticonvulsivos y el nombramiento de su dosis efectiva también deben ser manejados por un médico especialista. También decide si habrá suficiente de un medicamento o si el paciente tendrá que tomar 2 o más medicamentos durante un tiempo prolongado .
Para la mayoría de los pacientes, los médicos prescriben preparaciones de ácido valproico (ácido valproico, Depakin, Convulex, valparina, etc.), que previenen las convulsiones, mejoran el estado de ánimo y el estado mental de los pacientes.
El ácido valproico está disponible en forma de tabletas, jarabes y soluciones inyectables. El medicamento más popular es el medicamento de acción prolongada "Depakin" en tabletas y como una solución para administración intravenosa. La dosificación de la droga la determina el médico individualmente según el peso, la edad y el estado del paciente.
Tome la droga durante las comidas de 2 a 3 veces al día. La dosis diaria promedio es de 20-30 mg por 1 kilogramo del peso del paciente, el máximo es de 50 mg / kg por día.
Contraindicaciones No se utiliza para las violaciones del hígado y el páncreas, diátesis hemorrágica, hepatitis, porfiria e hipersensibilidad a la droga.
Entre los efectos secundarios se pueden distinguir el temblor de manos, la digestión y las heces, cambios en el peso corporal.
"Topiramate" es también una droga de elección en California. Está hecho en forma de tabletas y se usa como parte de la monoterapia y en combinación con otros medicamentos.
Método de aplicación y dosificación. Tome las pastillas adentro sin importar la ingesta de alimentos. La ingesta diaria inicial para adultos es de 25-50 mg, para niños de 0.5-1 mg / kg. Cada semana, la dosis aumenta de acuerdo con la prescripción del médico.
El medicamento no debe tomarse durante el embarazo y la lactancia, así como con una mayor sensibilidad a sus componentes. La medicina tiene muchos efectos secundarios diferentes.
Medicamentos que el médico puede prescribir en el síndrome de Angelman "Klomazepam", "Rivotril" "lamotrigina", "Seyzar", "Lamictal", "levetiracetam", "Keppra", "Epiterra" et al.
Tratamiento alternativo y homeopatía
La medicina alternativa, como los medicamentos homeopáticos, ciertamente difieren de la seguridad comparativa, pero aquí la efectividad de dicho tratamiento en relación con el síndrome de Angelholm puede considerarse controvertida.
Aunque algunos tratamientos alternativos todavía pueden ayudar. Se trata de detener las convulsiones epilépticas. En este sentido, la terapia a base de hierbas puede ser bastante efectiva.
Un buen efecto es proporcionado por una tarifa médica basada en peonía, regaliz y lenteja de agua (los componentes se toman en cantidades iguales). Los pastos deben ser molidos en harina. Después de 2 semanas desde el comienzo de la recepción, puede notar una disminución significativa en la frecuencia de ataques convulsivos.
Útil para los calambres y una decocción de lavanda (1 cucharadita de un vaso de agua hirviendo). La formulación se hierve durante 5 minutos e insistió durante media hora. Tome la medicina durante la noche por 14 días.
Efectivo para ataques epilépticos se considera agripalma de agua (o alcohol).
De medicamentos de homeopatía para prevenir las convulsiones con el síndrome de Angelman se puede utilizar medicamentos a base de manzanilla y motherwort, hydrocyanicum Acidum, Argentum nitricum, bromatum Kalium, Arsenicum album. Pero es necesario tener en cuenta que las dosis efectivas y seguras de los preparados en cada caso concreto pueden designar solamente al médico el homeópata.
Prevención
Como el lector probablemente ya haya entendido, para prevenir la mutación de los genes y otras anormalidades cromosómicas, la medicina todavía está más allá del poder, sin embargo, así como para remediar la situación. Esto puede sucederle a todos, porque los niños con síndrome de Angelmann nacen también en padres sanos, y la genética, que en este momento es una de las ramas menos estudiadas de la medicina, todavía no puede explicar esto.
Lo único que se puede hacer es responsabilizarse responsablemente de la planificación del embarazo, registrarse y examinarse a tiempo. Pero, de nuevo, tal medida no sería profiláctica, sino cognitiva, como cualquier encuesta. Pero los padres jóvenes de antemano sabrán para qué prepararse y, en caso de una respuesta positiva, decidirán si podrán asumir la responsabilidad de criar a un niño enfermo.
Pronóstico
El pronóstico para el síndrome de Anghelman depende de la naturaleza de la anomalía cromosómica y la oportunidad de su detección. La parte más difícil es para aquellos niños cuyos 15 cromosomas contienen genes "perdidos" (supresión). La probabilidad de caminar y hablar en tales pacientes es extremadamente pequeña. Los casos restantes con un enfoque atento y amor por su hijo son susceptibles de corrección.
Tales pacientes, por desgracia, no pueden convertirse en miembros plenos de la sociedad, a pesar del hecho de que están lejos de ser estúpidos, entienden el habla y su significado. Aquí hay problemas con la comunicación que tienen para la vida. A los pacientes se les puede enseñar lenguaje de señas desde la infancia, pero a uno no se le puede obligar a comunicarse con palabras. El léxico de los pacientes que "hablan" se limita a un mínimo de palabras utilizadas en la vida cotidiana (5-15 palabras).
En cuanto a la esperanza de vida y la salud general de los pacientes con síndrome de Anghelman, las cifras aquí fluctúan en promedio. En la edad adulta, los pacientes generalmente enfrentan problemas de salud como escoliosis y obesidad, que, con el enfoque correcto para el tratamiento, no son potencialmente mortales.