Médico experto del artículo.

Nuevos artículos
Hamartoma
Último revisado: 07.06.2024

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
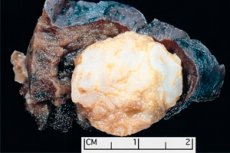
La formación de tipo tumor localizada en cualquier región anatómica resultante del crecimiento anormal del tejido benigno, en medicina se define como hamartoma (del griego hamartia - error, defecto). [1]
Epidemiología
Estadísticamente, los hamartomas representan el 1.2% de las neoplasias benignas. La prevalencia de hamartomas pulmonares se estima en aproximadamente el 0.25% de la población general y representa hasta el 8% de todas las neoplasias pulmonares. La mayoría de los hamartomas pulmonares se diagnostican incidentalmente en pacientes de 40 a 70 años de edad, pero son muy raros en la práctica pediátrica.
En general, la mayoría de los hamartomas se diagnostican en hombres, aunque en el riñón son más comunes en las mujeres y se identifican en la mediana edad.
Alrededor del 5% de los tumores de mama benignos son hamartomas, y más comúnmente afectan a las mujeres mayores de 35 años.
El 80-90% de las lesiones hamartomatosas del cerebro y más del 50% de los hamartomas del corazón están asociadas con la esclerosis tuberosa.
Causas Hamartomas
Los gamartomas pertenecen a malformaciones congénitas y son formaciones de carácter benigno, que se forman a partir de tejidos mesenquimales que se originan en las láminas germinales. Y las causas de su ocurrencia están asociadas con la división celular no controlada de los tejidos citológicamente normales (músculo conectivo, liso, grasa o cartílago), característicos de una ubicación anatómica dada y su sobrecrecimiento focal durante la embriogénesis de casi cualquier órgano o estructura anatómica.
La aparición de hamartomas múltiples en el mismo paciente a menudo se conoce como hamartomatosis o hamartoma pleiotrópico.
Estos tumores pueden ocurrir esporádicamente o en presencia de ciertas enfermedades hereditarias dominantes autosómicas, así como síndromes determinados genéticamente.
En muchos casos, los hamartomas se forman cuando una enfermedad genética rara de la naturaleza multisistémica - esclerosis tuberosa -se manifiesta poco después del nacimiento, o en la enfermedad familiar de Recklinghausen - neurofibromatosis tipo 1. [2]
Factores de riesgo
Los principales factores de riesgo para la formación de hamartoma incluyen la presencia de los llamados síndromes genéticos de la poliposis hamartomatosa en la historia de los pacientes, que incluyen:
- Síndrome de hamartoma múltiple- Síndrome de Cowden, en el que se forman múltiples hamartomas de origen ecto, ento y mesodérmico, se observan poliposis gastrointestinal y manifestaciones mucocutáneas;
- Síndrome proteico;
- Síndrome de Weil - poliposis juvenil del colon;
- El síndrome de Bannayan-Riley-Ruvalcaba, que, como el síndrome de Cowden, produce múltiples hamartomas (pólipos hamartomatosos) del intestino;
- Síndrome de Carney-Stratakis y complejo Carney.
Además, los hamartomas se forman en pacientes con síndrome hereditario de Watson y en casos de síndrome de palanca de palanca esporádica o congénita con hamartoma hipotalámico y polidactilia.
Patogenesia
El mecanismo de mayor proliferación de tejidos germinales con la formación de malformaciones de tipo tumoral en varios órganos se explica por aberraciones cromosómicas y mutaciones genéticas que pueden ocurrir espontáneamente o ser heredadas.
En la esclerosis tuberosa, se han identificado mutaciones en los genes TSC1 o TSC2: supresores tumorales que prevengan e inhiben la proliferación excesiva, se han identificado un crecimiento y división celular demasiado rápido o no controlado. Y en la neurofibromatosis tipo 1 y el síndrome de Watson - mutaciones de línea germinal del gen supresor tumoral mitocondrial NF1.
En el síndrome tumoral de Hamartoma, que combina los síndromes de Cowden, Protea, Bannayan-Riley-Ruvalcaba y Polyiposis juvenil, la patogénesis se asocia con la mutación del gen PTEN, que codifica una enzima involucrada en la regulación de la proliferación y se considera un gen supresor tumoral.
Las mutaciones en el gen STK11 que codifican la estructura y la función de una de las enzimas de serina transmembrana, lo que reduce su capacidad para restringir la división celular, conduce al síndrome de Peutz-Jeghers-Turen, con el desarrollo de pólipos intestinales y lesiones cutáneas pigmentadas. Se ha identificado una mutación en el gen GLI3, un factor de transcripción involucrado en la formación de tejido intrauterino, en el síndrome de Pallister-Hall.
Por lo tanto, el crecimiento celular no controlado debido a mutaciones genéticas conduce a la formación de hamartoma.
Síntomas Hamartomas
Dependiendo de la localización de los hamartomas, sus tipos se distinguen, y cada uno de ellos tiene su propia estructura y sintomatología.
Hamartoma del pulmón
El hamartoma pulmonar se puede formar en cualquier parte del lóbulo y periférico de los pulmones y consiste en tejidos normales presentes en los pulmones: adiposo, epitelial, fibroso y cartilaginoso. En el 80% de los casos, el componente condroide (células de cartílago hialino) predomina con la inclusión de adipocitos: células de tejido adiposo y células epiteliales de las vías respiratorias. [3]
Los nombres anteriores: Hamartoma Chondroid, Mesenchymoma, Hamartoma condomatoso o Hamartocondroma no son recomendados actualmente por la OMS.
El hamartoma quístico mesenquimatoso del pulmón, por otro lado, es menos común y se asocia con el síndrome de Cowden en la mayoría de los pacientes.
La lesión hamartomatosa del pulmón puede no manifestarse, pero puede causar síntomas en forma de tos crónica (a menudo con hemoptisis), sibilantes al respirar y dificultad para respirar. [4]
Un hamartoma del corazón
Los tumores benignos primarios tumores cardíacos en adultos incluyen hamartoma de miocito maduro, y en bebés y niños con esclerosis tuberosa, rabdomioma, es decir, hamartoma miocárdico de los ventrículos o tabique interventricular. [5]
El hamartoma de cardiomiocito maduro se desarrolla en la pared ventricular (y rara vez en las aurículas) y puede aparecer como múltiples lesiones que son masas densas estrechamente asociadas con el miocardio subyacente. El tumor puede causar síntomas de insuficiencia cardíaca: dolor en el pecho, palpitaciones y arritmias, soplos cardíacos, edema, disnea, cianosis.
Los rabdomiomas cardíacos, la mayoría de los cuales se diagnostican dentro del primer año de vida, están compuestos de tejido muscular cardíaco formado por mioblastos embrionarios y tienen la aparición de masas focales sólidas sin una cápsula.
Típicamente, estos hamartomas presentan asintomáticamente y espontáneamente retrocediendo antes de los 4 años.
Las lesiones hamartomatosas también son consideradas por algunos expertos asociados con el complejo Carney myxoma del corazón. [6]
Gamartoma del tracto gastrointestinal
El hamartoma gástrico es una masa mesenquimatosa en forma de un pólipo epitelial hiperplásico del estómago, pólipo Peutz-Jeghers y un raro hamartoma mioepitelial-con lágrimas de músculo liso hipertófiadas. Otros nombres para este hamartoma incluyen hamartoma mioglandular, hamartoma adenomatomatoso y adenomio gástrico. Las manifestaciones clínicas típicas incluyen dispepsia, dolor epigástrico y sangrado gastrointestinal superior. [7], [8]
Más información en el material - poliposis gástrica
Un hamartoma intestinal es un hamartomatoso o hiperplásico pólipo del intestino grueso, diagnosticado como un adenoma adenomatoso o tubular. Cuando el hamartoma se localiza en la glándula de Brunner del duodeno, los síntomas se manifiestan por el dolor en la región epigástrica; náuseas, vómitos y flatulencia (que indican obstrucción intestinal); y, si de tamaño considerable, sangrado gastrointestinal. En casos de hamartoma mioepitelial del íleon, los pacientes se quejan de dolor abdominal, han disminuido el peso corporal y desarrollan anemia crónica. [9], [10]
También lea - pólipos rectales
El hamartoma retrorectal es un hamartoma quístico o un quiste multicambo del espacio retrorectal (el tejido conectivo suelto entre el recto y su propia fascia) que ocurre más comúnmente en mujeres de mediana edad. Tiene la apariencia de un quiste que sale de la pared posterior del recto, que está revestida con epitelio y contiene fibras de músculo liso dispuestos caóticamente. Este hamartoma se presenta con dolor abdominal más bajo y estreñimiento recurrente. [11], [12]
Hamartomas del hígado y el bazo
El hamartoma biliar múltiple del hígado es un hamartoma de los conductos biliares intrahepáticos interdollic asociados con malformaciones de su desarrollo durante el período embrionario. Este hamartoma (único o múltiple) consiste en grupos de conductos biliares y dilatados al azar y el estroma fibrocolagenoso. [13]
Los hamartomas biliares son asintomáticos y generalmente se descubren incidentalmente (durante el examen radiológico o la laparotomía). [14]
Una neoplasia primaria rara y a menudo detectada por cierto es un hamartoma del bazo, que consiste en elementos de la pulpa roja del bazo, en forma de una masa homogénea bien definida de consistencia firme. Esta malformación puede ser única o múltiple; Al exprimir el parénquima esplénico, puede haber una sensación de incomodidad y dolor en el área subcostal izquierda. [15], [16]
Hamartomas renales
El hamartoma más común del riñón se diagnostica como angiomiolipoma del riñón, ya que este tumor benigno consiste en tejido adiposo maduro con fibras musculares lisas incrustadas y vasos sanguíneos. Se forma en la esclerosis tuberosa en el 40-80% de los casos. El aumento del tamaño del hamartoma (más de 4-5 cm) conduce al dolor y la apariencia de sangre en la orina. [17], [18]
Hamartoma del pecho
Las definiciones de diagnóstico de la OMS de hamartoma mama son términos como adenolipoma, condrolipoma y hamartoma mioide. Aunque a menudo llamado fibroadenolipoma por mammólogos, porque la formación de tumores contiene células de tejido fibroso, glandular y adiposo encerrado en una cápsula delgada de tejido conectivo con contornos distintos. Se pueden observar calcificaciones focales en la visualización. En este caso, las manifestaciones clínicas están ausentes. [19], [20]
También lea - tumores de mama
Hamartomas del cerebro
Un tercio de los pacientes con esclerosis tuberosa tienen un hamartoma cerebral en forma de crecimiento cortical intracraneal o tubérculos en varios lóbulos, en el borde de la materia gris y blanca, o nódulos subependimales a lo largo de las paredes de los ventrículos cerebrales. El hamartoma astrocítico, un astrocitoma de células gigantes subependimales con interrupción cortical, neuronas dismórficas y grandes células gliales del parénquima cerebral (astrocitos), también pueden formarse. Los síntomas de los hamartomas cerebrales incluyen ataques de convulsiones y retraso mental en los niños. [21], [22]
Una malformación rara que ocurre durante la embriogénesis y está presente al nacer es un hamartoma hipotalámico, que es una masa de neuronas heterotópicas y células gliales. A medida que el cerebro del niño crece, el tumor se agranda pero no se propaga a otras regiones cerebrales. [23], [24]
Si se forman tejidos hipertrofiados en la parte anterior del hipotálamo (tuber cinereum), donde la glándula pituitaria se adhiere a ella, la malformación manifiesta los síntomas de los síntomas centrales desarrollo sexual prematuro (antes de 8-9 años de edad): la aparición de acné, el desarrollo temprano, el desarrollo temprano de las glands de las mamás y las mujeres tempranas en las mujeres tempranas; Early Pur Púbico y Mutación de voz en niños.
When hamartomas form in the posterior part of the hypothalamus, there may be abnormalities in the electrical activity of the brain, which in early infancy is manifested by seizures, and at a later stage (ages 4 to 7 years) by epilepsy with focal epileptic seizures with sudden laughter or with involuntary crying, atonic and tonic-clonic seizures, as well as seizures of aggression, memory and cognitive problemas.
Un hamartoma pituitario es un benigno esporádico adenoma pituitario.
Los adultos de mediana edad con síndrome de Cowden pueden tener una masa rara de tipo tumor, un hamartoma del cerebelo, diagnosticado como gangliocitoma cerebeloso displásico o enfermedad de Lhermitte-Duclos. Los síntomas pueden estar ausentes o manifestados como dolor de cabeza, mareos, coordinación deteriorada de los movimientos y parálisis de los nervios craneales individuales.
Hamartoma de ganglio linfático
Cuando las células del músculo liso y el tejido adiposo, así como los vasos sanguíneos y el estroma colágeno de los ganglios linfáticos inguinales, retroperitoneales, submandibulares y cervicales sobrecargados, un hamartoma angiomatoso angiomatoso de un hamartoma angiomatoso o nodomatoso de un ganglio linfático o un hamartoma angeomatoso nodo, con un hamartoma parcial o completo de su parénquima. [25], [26]
Un hamartoma de la piel
En presencia de esclerosis tuberosa o neurofibromatosis, se observan varios hamartomas de la piel, con mayor frecuencia en forma de manchas hipopigmentadas; Café y manchas de leche;
Una manifestación dermatológica rara de la esclerosis tuberosa (especialmente en los hombres) es el hamartoma foliculocíntico y de colágeno, caracterizado por abundante deposición de colágeno en la dermis, la fibrosis perifolicular concéntrica y la exámenes histopatológicos concéntricos en forma de embudo de queratina observadas en el examen histopatológico. [27]
Para los hamartomas que consisten en melanocitos (células que producen la melanina pigmentaria), la mayoría de los expertos también se refieren a varios neoplasias melanocíticas, en particular nevos melanocíticos congénitos, que representan una abnormalidad de la embiogenesis.
En términos de etiología, los hamartomas compuestos de tejido vascular también son hemangiomas de la piel.
Los pacientes con síndrome de Peutz-Jeghers-Thuren tienen hamartoma en forma de pigmentación irregular de la piel y membranas mucosas - lentiginosis periorificialis
Los casos de hamartoma ectodérmico-mesodérmico papular lineal (Hamartoma moniliformis) muestran una erupción papular lineal de color carne en la cabeza, el cuello y la parte superior del pecho.
Y un hamartoma sebocítico es un hamartoma de las glándulas sebáceas, lea más en la publicación - nevus sebáceo.
Hamartoma del ojo
Las lesiones hamartomatosas pigmentadas del iris en la neurofibromatosis tipo 1 y el síndrome de Watson, en forma de grupos nodulares de melanocitos dendríticos, se definen como hamartomas de iris o nódulos de lisch. Son las pápulas de marrón amarillo en forma de cúpula transparente (generalmente no afectan) que sobresalen sobre la superficie del iris.
Y pacientes con angiofibroma juvenil de la nasofaringe y poliposis adenomatosa familiar a menudo desarrollan hamartoma combinado de la retina y el epitelio de pigmento retiniano-en forma de una mancha negra en la parte central (macular) de la retina. [28]
Un hamartoma de la nariz
El hamartoma nasal se define por los especialistas como hamartoma nasal condromesenquimal o condroma nasal, debido a la proliferación benigna del epitelio respiratorio, las glándulas submucosas y el mesenquima condal-hueso. Sus manifestaciones clínicas dependen del tamaño y la localización de la lesión e incluyen: congestión nasal, dificultad en la respiración nasal y la lactancia materna en bebés, secreción nasal clara y sangrado nasal. Un hamartoma puede crecer con el niño y extenderse a las órbitas oculares, lo que resulta en el desplazamiento hacia adelante o hacia atrás del globo ocular, el estrabismo u alteraciones oculomotoras. [29]
Un hamartoma en un niño
Todas las lesiones hamartomatosas mencionadas anteriormente de varios órganos y estructuras anatómicas están presentes en niños con síndromes correspondientes.
Los recién nacidos se presentan con hamartoma mesenquimatoso de la pared torácica o hamartoma cartilaginoso de la costilla, que son masas inmóviles sólidas resultantes del sobrecrecimiento focal de los elementos esqueléticos normales con elementos cartilaginosos, vasculares y mesenquimales. Este hamartoma puede causar insuficiencia respiratoria y el desarrollo del síndrome de dificultad respiratoria. El hamartoma mesenquimal del hígado es el segundo más frecuente tumor de hígado benigno en niños. Esta formación similar al tumor (más a menudo localizada en el lóbulo derecho del órgano) consiste en células de estroma mesenquimatoso, hepatocitos y células epiteliales del revestimiento del conducto biliar. La imagen clínica incluye masa palpable en la cavidad abdominal, la anorexia y la pérdida de peso, y en caso de tamaño significativo (hasta 10 cm y más), el tumor cubre conductos biliares extrahepáticos y cava de vena inferior, que conduce a la ictericia y edema de las extremidades inferiores.
Un hamartoma es un nefroma mesoblástico congénito (que ocurre en 1 de cada 200,000 bebés) que puede provocar hinchazón abdominal en el recién nacido con una masa palpable de consistencia densa en el cuadrante superior derecho del abdomen. Los bebés también pueden presentarse con una respiración rápida y poco profunda.
Las anomalías congénitas raras incluyen hamartoma fibroso de infancia, que ocurre en niños en los primeros dos años de vida y se presenta como una masa nodular indolora en los tejidos subcutáneos de la axilla, el cuello, el hombro y el antebrazo, la espalda y el pecho, el muslo, el pie y el genitales externos.
El hamartoma angiomatoso eccrino en un niño puede estar presente al nacer o manifiesto en la primera infancia. Este tumor benigno de la naturaleza hamartomatosa generalmente tiene la apariencia de nódulos y/o placas azuladas o parduzco que resultan de la proliferación del tejido y los capilares de la glándula ecrina en las capas medias y profundas de la dermis. Este hamartoma puede causar hiperhidrosis localizada y un aumento del crecimiento del cabello.
Complicaciones y consecuencias
En general, se acuerda que los hamartomas rara vez se repiten o se transforman en tumores malignos. A menudo muestran poco o ningún síntoma y, a veces, incluso desaparecen con el tiempo. Pero en casos más severos y dependiendo del sitio de formación, estas malformaciones pueden tener complicaciones y consecuencias graves.
En primer lugar, un hamartoma puede crecer de tal tamaño que comienza a presionar sobre tejidos y órganos circundantes, lo que interrumpe sus funciones.
El hamartoma cardíaco en los niños puede conducir a anormalidades persistentes del ritmo cardíaco, defectos de la válvula y un flujo sanguíneo intracardíaco deteriorado con insuficiencia cardíaca congestiva posterior.
Las complicaciones de los pólipos hamartomatosos del tracto gastrointestinal son el sangrado gastrointestinal, la obstrucción y la intestinal intestinal (con posible resultado fatal). Y un gran hamartoma renal puede provocar ruptura del riñón.
Un hamartoma en el cerebro puede causar síndrome obstructivo de hidrocefalia.
En los hamartomas hipotalámicos y pituitarios, la producción de hormona somatotrópica (hormona del crecimiento) puede verse afectada, lo que lleva al desarrollo de nanismo hipofisario (hipopituitarismo) en niños. Los hamartomas hipotalámicos en niños también pueden conducir a la epilepsia resistente a las drogas.
Las complicaciones del hamartoma del epitelio del pigmento retiniano están cargados de disfunción del nervio retiniano y/u óptico, edema macular, neovascularización del coroides y desprendimiento de retina.
Diagnostico Hamartomas
Una parte importante del diagnóstico de hamartomas y síndromes relacionados es la recolección de anamnesis, incluida la historia familiar.
Las pruebas de laboratorio incluyen análisis de sangre: clínica general; electrolitos séricos; perfil de linfocitos; niveles de calcio, potasio, fosfato y urea; y pruebas de función hepática. Si es posible, se realiza una biopsia de punción de aspiración de aguja fina de la masa, ya que el examen histológico es crucial en el diagnóstico y elección de tácticas de tratamiento.
El diagnóstico instrumental proporciona la visualización de la formación e identificación de tumores hamartomatosos de su localización exacta, para la cual se utilizan rayos X, angiografía, electroencefalografía (EEG), ultrasonido (ecografía), TC (tomografía computarizada), PET (tomografía de emisión de positrones), MRI (resonancia magnética).
Diagnóstico diferencial
En cualquier masa anormal, el diagnóstico diferencial es muy importante. Por lo tanto, el tuberculoma y el hamartoma están diferenciados; Hamartoma pulmonar y cáncer de pulmón primario, carcinoide broncogénico, enfermedad metastásica. El hamartoma cerebral debe distinguirse del craneofaringioma y el glioma hipotalámico-quiasmático. Y el diagnóstico diferencial de hamartoma como nefroma mesoblástico congénito incluye tumor Wilms (nefroblastoma maligno), sarcoma de células claras del riñón y el tumor renal osificador en los bebés.
¿A quién contactar?
Tratamiento Hamartomas
Si el hamartoma es asintomático y se descubre accidentalmente, no se requiere tratamiento, pero es necesario monitorear su "comportamiento" y la condición del paciente. En otros casos, la terapia tiene como objetivo reducir la intensidad de los síntomas y prevenir complicaciones. Por ejemplo, en el hamartoma hipotalámico con síntomas de pubertad prematura, se prescriben ciertos medicamentos que inhiben la liberación de ciertas hormonas. Los medicamentos cardíacos se utilizan para tratar los síntomas de la insuficiencia cardíaca en pacientes con hamartomas cardíacos.
La eliminación quirúrgica de los hamartomas está indicada para confirmar el diagnóstico y en casos de síntomas intensos médicamente incorrectos.
Por ejemplo, los hamartomas pulmonares pueden resecarse mediante resección de cuña y, en casos severos, mediante la eliminación de un lóbulo del pulmón (lobectomía). También se puede extirpar un hamartoma mama, y si es grande, se puede requerir una mastectomía parcial o completa.
La termoablación de radiofrecuencia estereotáctica o la ablación con láser se pueden usar para eliminar los pólipos hamartomatosos. También se usa la radiocirugía con rayos gamma altamente enfocados (cuchillo gamma para hamartomas hipotalámicos o hamartomas astrocíticos.
Prevención
El único método para prevenir el desarrollo de hamartomas puede considerarse detección genética de los futuros padres del niño.
Pronóstico
El pronóstico general de esta anomalía congénita depende de la localización y el tamaño de la neoplasia, así como de las comorbilidades y la salud general del paciente.