Médico experto del artículo.

Nuevos artículos
Atresia de coanas
Último revisado: 07.06.2024

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
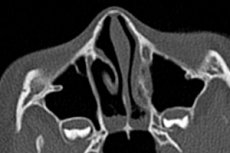
La ausencia completa de aberturas naturales en varias estructuras anatómicas del cuerpo se llama atresia (desde la negación griega de algo, tesis - apertura). Choanal atresia significa la ausencia de aberturas emparejadas en la parte posterior del pasaje nasal: pasajes nasales posteriores, que conectan la cavidad nasal con la nasofaringe. [1]
Epidemiología
La frecuencia de esta malformación es un caso por 5-8 mil nacimientos vivos (según otros datos, tres casos por 10 mil), y en el 65% de los casos, los niños nacen con atresia choanal unilateral.
Al mismo tiempo, en el 60-75% de los casos de atresia bilateral, los recién nacidos tienen malformaciones concomitantes: otras anomalías craneofaciales. Además, las estadísticas observan casi el 8% de los casos familiares.
Según algunos informes, hasta el 30% de los pacientes tienen la atresia choanal a cargo del síndrome. [2]
Causas Atresia de coanas
Como Choa Atresia en un niño recién nacido es una patología congénita, sus causas están relacionadas con la interrupción de las estructuras nasales durante el período embrionario del desarrollo intrauterino. Como resultado de estos trastornos, un tabique óseo/cartilaginoso o, más raramente, una membrana fibrosa (tejido conectivo) permanece entre la cavidad nasal (cavum nasi) y la parte superior de la nasofaringe (pars nasal faryarngis).
The genetic factor should be taken into account, especially in the presence of a complex of developmental defects, such as congenital CHARGE syndrome or cHARGE-association - with anomalies of the eye membranes, auricles, esophagus, genitalia, etc. The congenital syndromes of craniofacial (craniofacial) dysostosis or craniosyntosis (premature Fusión de una o más suturas craneales) causada por mutaciones genéticas, en la que existe una anomalía de la nasofaringe y los pasajes nasales posteriores, también incluyen síndrome de Treacher Collins; Alfie, DiGeorgi, Síndromes de Apert; síndrome edwards; Crouzon, Antley-Bixler, Pfeiffer, Tessier, Beer-Stevenson, Jackson-Weiss Síndromes; síndrome de alcohol fetal (síndrome de alcohol fetal).
Al deformar la poliposis nasal se desarrolla en niños y adultos jóvenes hay estenosis Choanal, es decir, su estrechamiento anormal, que se puede definir como estrechamiento de las vías respiratorias nasales en la región choanal posterior, la estenosis nasofarilgae o la atesia del choanal parcial.
Por lo tanto, en la otorrinolaringología, la atresia choanal adquirida (estenosis secundaria anterior y posterior de la cavidad nasal con la formación de septos fibrosos) a menudo también se reconoce. Esta condición puede ser el resultado de la sífilis, el lupus eritematoso sistémico, trauma a los senos paranasales, intervención quirúrgica, así como una consecuencia de la radiaterapia para tumores malignos de la nasofaringnx.
Sin embargo, la atresia Choanal es clasificada por los expertos médicos como una patología congénita, y practicar otorrinolaringólogos debe distinguirlo de la estenosis de los pasajes nasales posteriores, lo que no resulta en una obstrucción completa.
La atresia unilateral es dos veces más común: atresia choanal del lado derecho o atresia choanal del lado izquierdo, respectivamente. [3]
Factores de riesgo
Además de las anormalidades genéticas, varias exposiciones embriotóxicas y factores ambientales se reconocen como factores de riesgo para la atresia de la nasal distal del agujero.
Por lo tanto, un mayor riesgo de esta anomalía en el feto puede estar expuesto a las futuras madres que han tomado medicamentos del grupo de tioamida en hipertiroidismo durante el embarazo (para reducir el nivel de hormonas tiroideas). En tales casos, el embrión puede carecer de hormonas tiroideas, lo que afecta negativamente la morfogénesis de los órganos respiratorios superiores.
Además, los estudios han encontrado una posible asociación de atresia choanal neonatal con altas dosis de vitamina B12, B3 (PP), D y zinc durante el embarazo. El alcohol, el humo del tabaco y la cafeína tienen un impacto extremadamente negativo en el desarrollo de las estructuras craneofaciales del feto. [4]
En 2010-12, se informó un aumento en los nacimientos con atresia Choanal en los Estados Unidos debido a la exposición de las mujeres embarazadas a los productos químicos utilizados para tratar los cultivos.
Leer más:
Patogenesia
Choans (Latin. Choane (Latin: Funnel) son aberturas que conducen desde la cavidad nasal en la nasopharynx, limitadas en el medio por el enchufe (borde de la placa de hueso); hueso palatino (su placa horizontal).
La formación de Choanae, que se origina en los arcos branquiales del embrión, comienza en la cuarta semana de gestación (y continúa hasta la octava semana) con la migración de las células neurales de la cresta en los pliegues neuronales dorsales. A continuación, el pliegue epitelial dispuesto verticalmente (membrana oronasal) entre el techo de la cavidad oral primaria y los procesos nasales (Placoda nasal) en la superficie lateral de las rupturas de la cabeza. Los procesos nasales se profundizan en el mesodermo, lo que conduce a la formación de fosa nasales y luego a la choanae primaria (primitiva).
Teóricamente, la patogénesis de la anomalía congénita de la atresia choanal puede deberse a la preservación de la membrana de la caríngea-faríngea (bucopofaringeal), una capa delgada de ectodermo y células entodermas que cubren la "apertura oral" del embrio por encima del final craneial del acorde. Esta membrana debería perforar en la sexta semana de gestación, pero por razones desconocidas puede no, lo que resulta en defectos orofaciales como el paladar hendido y la atresia Choanal.
También posible: preservación de la membrana bucconasal (una capa delgada de tejido epitelial, que debe reabsorbir en la séptima semana de gestación); adherencia anormal del tejido mesodérmico en el área de Choanas; Trastorno local de la migración de células mesenquimales a lo largo de la cresta neural, lo que lleva a defectos en la formación de la protuberancia frontonasal del embrión de la parte de la cabeza y sus ramificaciones.
Pero ninguno de los supuestos del mecanismo de desarrollo de la atresia nasal posterior no tiene evidencia hasta la fecha.
Síntomas Atresia de coanas
Los recién nacidos respiran por la nariz porque su epiglotis es más alto (en comparación con los adultos), y la laringe aumenta durante la deglución, tocando la nasofaringe, y se cierra entre el paladar blando y los lados de la nasofaringe. Y la capacidad de respirar por la boca aparece a las 4-6 semanas después del nacimiento, después de la disminución de la laringe.
Por lo tanto, los síntomas clásicos exhibidos por la atresia bilateral choanal en los recién nacidos se deben al deterioro completo de la función respiratoria.
Por ejemplo, el bebé tiene cianosis cíclica indicativa de episodios de asfixia: la lividez de la piel, que disminuye al llorar (cuando el niño abre la boca de par en par y respira dentro y fuera) y se repite tan pronto como el llanto se detiene y el bebé cierra la boca. En tales casos, se requiere atención médica de emergencia: intubación endotraqueal o traqueotomía.
La atresia unilateral (es decir, la ausencia de solo un pasaje nasal posterior) a menudo se detecta más tarde en la vida (a los 5-10 meses de edad o mucho más tarde) y sus primeros signos son unilaterales congestión nasal. Además, hay una descarga persistente de una fosa nasal - rhinorrea, ronquidos y estridor (respiración ruidosa), así como sinusitis crónica. [5]
Complicaciones y consecuencias
La atresia bilateral choanal conduce a la aguda síndrome de dificultad respiratoria neonatal debido a la obstrucción completa de las vías respiratorias nasales.
Consecuencias y complicaciones de la atresia unilateral: distorsión de proporciones faciales, crecimiento deteriorado de las mandíbulas superiores e inferiores y la formación de una mordida patológica, debido al desarrollo craneofacial inadecuado; La aparición de aPNEA DE NIGHTA obstructiva y otros problemas respiratorios asociados con el funcionamiento deteriorado del tracto respiratorio superior. [6]
Diagnostico Atresia de coanas
Si se sospecha la atresia bilateral neonatal choanal, un neonatólogo realiza el diagnóstico clínico preliminar en una emergencia insertando un tubo nasogástrico a través de la cavidad nasal del bebé. La sospecha de esta anomalía congénita se confirma si no se puede insertar el catéter.
Para confirmar el diagnóstico, las imágenes son necesarias: endoscopia (examen) de la cavidad nasal, tomografía computarizada de la nariz, senos paranasales y estructuras óseas paranasales.
La atresia choanal unilateral es la forma más común y puede no tener defectos de nacimiento asociados, por lo que puede no ser diagnosticada inmediatamente después del nacimiento.
En la atresia unilateral, también se realizan diagnósticos instrumentales: rinoscopia anterior y posterior; endoscopia de la cavidad nasal y tomografía computarizada nasal; rinomanometría - estudio de la función respiratoria nasal.
Diagnóstico diferencial
El diagnóstico diferencial incluye problemas de respiración nasal que pueden deberse a: tabique nasal desviado o dislocación de cartílago; estenosis de la cavidad nasal e hipertrofia congénita de los huesos nasales inferiores; estenosis aislada del agujero nasal en forma de pera (restricción ósea anterior del esqueleto nasal); un pólipo antrochoanal, quiste dermoide de la cavidad nasal o quiste del conducto nasolacrimal; Hemangioma o Garmatoma nasal.
¿A quién contactar?
Tratamiento Atresia de coanas
En el caso de la atresia Choanal, solo el tratamiento quirúrgico por resección endoscópica transnasal y la coanoplastia con TC o resonancia magnética previa de la cavidad nasal se realiza para restaurar su permeabilidad.
La intervención quirúrgica para la atresia bilateral choanal generalmente se realiza dentro de los primeros tres meses de vida, y en casos de atresia unilateral, después de que el niño tiene dos años. [7]
Todos los detalles en la publicación - restauración de Choanal Atresia
Prevención
Dados los factores de riesgo conocidos para este defecto de nacimiento, las medidas preventivas pueden considerarse un manejo adecuado del embarazo y la extrema precaución al prescribir cualquier medicamento a las futuras madres.
Y las parejas responsables tienen una manera de evitar tener un hijo con un síndrome genéticamente determinado, como el asesoramiento médico genético.
Pronóstico
La atresia bilateral choanal es potencialmente mortal para el recién nacido, pero con un tratamiento oportuno y efectivo y sin asociación con síndromes congénitos, el pronóstico generalmente se considera bueno.
Libros sobre Atresia de Choanae
- Otorrinolaringología pediátrica: principios y vías de práctica " - por Christopher J. Hartnick et al. (Año de lanzamiento: 2015)
- "Otorhinolaringología de Scott-Brown, cirugía de cabeza y cuello" - Autor: John C Watkinson et al. (Año de publicación: 2020)
- "Cummings Otolaryngology: Cirugía de cabeza y cuello" - Autor: Paul W. Flint et al. (Año de lanzamiento: 2020)
- "ENT: una introducción y guía práctica" - Autor: Sharan K. Naidoo (Año de la liberación: 2018)
Literatura utilizada
- Palchun, Magomedov, Alexeeva: Otorhinolaringología. Manual nacional. Geotar-Media, 2022.
- Atresia congénita choanal en niños. Libro de texto para estudiantes de medicina. Kotova E.N., Radtsig E.YU. 2021