Médico experto del artículo.

Nuevos artículos
Enfermedad de Charcot-Marie-Tooth.
Último revisado: 12.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
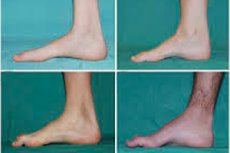
La atrofia muscular peronea, síndrome o enfermedad de Charcot-Marie-Tooth es un grupo de enfermedades hereditarias crónicas con daño a los nervios periféricos.
Según la CIE-10, en la sección de enfermedades del sistema nervioso, el código para esta enfermedad es G60.0 (neuropatía motora y sensitiva hereditaria). También se incluye en la lista de enfermedades huérfanas.
Epidemiología
Según las estadísticas clínicas, la prevalencia de todos los tipos de enfermedad de Charcot-Marie-Tooth por cada 100 mil habitantes es de 19 casos (según otras fuentes, un caso por cada 2,5-10 mil habitantes).
La CMT tipo 1 representa aproximadamente dos tercios de los casos (un caso por cada 5000 a 7000 habitantes), y casi el 70 % de ellos se asocian con la duplicación del gen PMP22. Más de 1,2 millones de personas en todo el mundo padecen este tipo de enfermedad.
La incidencia de CMT tipo 4 se estima en 1 a 5 casos por cada 10.000 niños. [ 1 ]
Causas Enfermedad de Charcot-Marie-Tooth
Según la clasificación de los síndromes polineuropáticos, la atrofia muscular peronea (peronea), amiotrofia neural de Charcot-Marie-Tooth o enfermedad de Charcot-Marie-Tooth (abreviada como CMT) se refiere a polineuropatías motoras-sensitivas determinadas genéticamente. [ 2 ]
Es decir, las causas de su aparición son mutaciones genéticas. Dependiendo de la naturaleza de las anomalías genéticas, se distinguen los principales tipos de este síndrome: desmielinizante y axonal. El primer grupo incluye la enfermedad de Charcot-Marie-Tooth tipo 1 (CMT1), que se produce debido a la duplicación del gen PMP22 en el cromosoma 17, que codifica la proteína transmembrana periférica de la mielina 22. Como resultado, se produce una desmielinización segmentaria de la vaina axónica (prolongaciones de las células nerviosas) y una disminución de la velocidad de conducción de la señal nerviosa. Además, pueden existir mutaciones en otros genes.
La forma axonal es la enfermedad de Charcot-Marie-Tooth tipo 2 (CMT2), que afecta a los propios axones y se asocia con cambios patológicos en el gen MFN2, ubicado en el locus 1p36.22, que codifica la proteína de membrana mitofusina-2, necesaria para la fusión mitocondrial y la formación de redes mitocondriales funcionales en el interior de las células nerviosas periféricas. Existen más de una docena de subtipos de CMT2 (con mutaciones en genes específicos).
Cabe destacar que actualmente se han identificado más de cien genes cuyo daño, transmitido hereditariamente, causa diversos subtipos de la enfermedad de Charcot-Marie-Tooth. Por ejemplo, las mutaciones en el gen RAB7 causan CMT tipo 2B; la alteración del gen SH3TC2 (que codifica una de las proteínas de membrana de las células de Schwann) causa CMT tipo 4C, que se manifiesta en la infancia y se caracteriza por la desmielinización de las neuronas motoras y sensoriales (existen una docena y media de formas del tipo 4 de esta enfermedad).
Un tipo raro de CMT 3 (llamado síndrome de Dejerine-Sottas) comienza a desarrollarse en la primera infancia y es causado por mutaciones en los genes PMP22, MPZ, EGR2 y otros.
Cuando la enfermedad de CMT tipo 5 aparece entre los 5 y 12 años, no solo se observa neuropatía motora (en forma de paraparesia espástica de las extremidades inferiores), sino también daño a los nervios óptico y auditivo.
La debilidad muscular y la atrofia óptica (con pérdida de visión), así como los problemas de equilibrio, son típicos de la CMT tipo 6. Y en la enfermedad de Charcot-Marie-Tooth tipo 7 no solo hay neuropatía sensitivo-motora, sino también una enfermedad de la retina en forma de retinosis pigmentaria.
Más común entre los varones, la CMT ligada al cromosoma X o enfermedad de Charcot-Marie-Tooth con tetraparesia de las extremidades (debilidad de movimiento de brazos y piernas) es un tipo desmielinizante y se cree que es resultado de una mutación en el gen GJB1 en el brazo largo del cromosoma X, que codifica la conexina 32, una proteína transmembrana de las células de Schwann y oligodendrocitos que regula la transmisión de señales nerviosas. [ 3 ]
Factores de riesgo
El principal factor de riesgo para CMT son los antecedentes familiares de la enfermedad, es decir, en parientes cercanos.
Según los genetistas, si ambos progenitores son portadores del gen autosómico recesivo de la enfermedad de Charcot-Marie-Tooth, el riesgo de tener un hijo que la desarrolle es del 25 %. Y el riesgo de que el hijo sea portador de este gen (pero no presente síntomas) se estima en un 50 %.
En caso de herencia ligada al cromosoma X (cuando el gen mutado se encuentra en el cromosoma X de la mujer), existe un 50 % de riesgo de que la madre transmita el gen a su hijo, quien desarrollará CMT. La enfermedad puede no presentarse al nacer una niña, pero los hijos de la hija (nietos) podrían heredar el gen defectuoso y desarrollar la enfermedad.
Patogenesia
En cualquier tipo de enfermedad de Charcot-Marie-Tooth, su patogenia es causada por una anomalía hereditaria de los nervios periféricos: motor (movimiento) y sensitivo (sensitivo).
Si el tipo de CMT es desmielinizante, entonces la destrucción o defecto de la vaina de mielina que protege los axones de los nervios periféricos conduce a una desaceleración en la transmisión de los impulsos nerviosos en el sistema nervioso periférico - entre el cerebro, los músculos y los órganos sensoriales.
En el tipo axonal de la enfermedad, los propios axones se ven afectados, lo que afecta negativamente la fuerza de las señales nerviosas, que es insuficiente para la estimulación completa de los músculos y los órganos sensoriales.
Lea también:
¿Cómo se transmite el síndrome de Charcot-Marie-Tooth? Los genes defectuosos pueden heredarse de forma autosómica dominante o autosómica recesiva.
El tipo más común, la herencia autosómica dominante, se presenta cuando existe una copia del gen mutado (portado por uno de los progenitores). La probabilidad de transmitir CMT a cada hijo se estima en un 50 %. [ 4 ]
En la herencia autosómica recesiva, se necesitan dos copias del gen defectuoso (una de cada padre que no muestre ningún signo de la enfermedad) para que la enfermedad se desarrolle.
En el 40-50% de los casos se produce desmielinización hereditaria autosómica dominante, es decir, CMT tipo 1; en el 12-26% de los casos, CMT axonal, es decir, tipo 2. Y en el 10-15% de los casos se observa herencia ligada al cromosoma X. [ 5 ]
Síntomas Enfermedad de Charcot-Marie-Tooth
Por lo general, los primeros signos de esta enfermedad comienzan a aparecer en la infancia y la adolescencia y se desarrollan gradualmente a lo largo de la vida, aunque el síndrome puede manifestarse más adelante. La combinación de síntomas es variable, y la velocidad de progresión de la enfermedad, así como su gravedad, son imposibles de predecir.
Los síntomas típicos de la etapa inicial incluyen mayor fatiga general; disminución del tono (debilidad) de los músculos de los pies, tobillos y espinillas; y falta de reflejos. Esto dificulta el movimiento del pie y provoca disbasia (alteración de la marcha) que se manifiesta por una mayor elevación de las piernas, a menudo con tropiezos y caídas frecuentes. Los signos de la enfermedad de Charcot-Marie-Tooth en niños pequeños pueden incluir torpeza pronunciada y dificultades para caminar, atípicas para la edad, asociadas con caída del pie bilateral. Las deformidades del pie también son características: arco alto (pie hueco) o pie plano severo, dedos curvados (en forma de martillo).
En el caso de caminar de puntillas en un contexto de hipotonía muscular, el neurólogo puede sospechar que el niño padece CMT tipo 4, en la que los niños pueden no ser capaces de caminar en la adolescencia.
A medida que la enfermedad progresa, la atrofia muscular y la debilidad se extienden a las extremidades superiores, lo que dificulta la motricidad fina y la realización de tareas manuales normales. La disminución de la sensibilidad táctil y de la capacidad para percibir calor y frío, así como el entumecimiento en pies y manos, indican daño a los axones de los nervios sensoriales.
En la enfermedad de Charcot-Marie-Tooth tipos 3 y 6 que se manifiesta en la infancia, se observan ataxia sensorial (alteración de la coordinación de movimientos y del equilibrio), espasmos musculares y temblores, daño al nervio facial, atrofia del nervio óptico con nistagmo y pérdida de audición.
En etapas posteriores, puede haber temblores incontrolables y calambres musculares frecuentes; problemas con el movimiento pueden llevar al desarrollo de dolor: muscular, articular, neuropático.
Complicaciones y consecuencias
La enfermedad de Charcot-Marie-Tooth puede tener complicaciones y consecuencias como:
- esguinces y fracturas más frecuentes;
- contracturas asociadas a acortamiento de músculos y tendones periarticulares;
- escoliosis (curvatura de la columna vertebral);
- Problemas respiratorios: cuando las fibras nerviosas que inervan los músculos del diafragma están dañadas:
- pérdida de la capacidad de moverse independientemente.
Diagnostico Enfermedad de Charcot-Marie-Tooth
El diagnóstico incluye examen clínico, anamnesis (incluidos antecedentes familiares), examen neurológico y sistémico.
Se realizan pruebas para evaluar el rango de movimiento, la sensibilidad y los reflejos tendinosos. La conducción nerviosa puede evaluarse mediante diagnósticos instrumentales como la electromiografía o la electroneuromiografía. También puede requerirse una ecografía o una resonancia magnética. [ 6 ]
Las pruebas genéticas o de ADN para detectar las mutaciones genéticas más comunes que causan CMT en una muestra de sangre son limitadas porque actualmente no existen pruebas de ADN para todos los tipos de la enfermedad. Para más información, consulte Pruebas genéticas.
En algunos casos, se realiza una biopsia del nervio periférico (generalmente el nervio sural).
Diagnóstico diferencial
El diagnóstico diferencial incluye otras neuropatías periféricas, distrofia muscular de Duchenne, síndromes mielopáticos y miasténicos, neuropatía diabética, mielopatías en esclerosis lateral múltiple y amiotrófica, síndrome de Guillain-Barré, traumatismo del nervio peroneo y su atrofia (incluso cuando se pinza entre los discos lumbares de la columna), daño al cerebelo o al tálamo, así como efectos secundarios de la quimioterapia (durante el tratamiento con citostáticos como vincristina o paclitaxel). [ 7 ]
¿A quién contactar?
Tratamiento Enfermedad de Charcot-Marie-Tooth
Hoy en día, el tratamiento para esta enfermedad hereditaria incluye terapia de ejercicios (dirigida a fortalecer y estirar los músculos); terapia ocupacional (que ayuda a los pacientes con debilidad muscular en las manos); y el uso de dispositivos ortopédicos para facilitar la marcha. Si es necesario, se recetan analgésicos o anticonvulsivos. [ 8 ]
En casos de pie plano severo, se puede realizar una osteotomía y, en caso de deformación del talón, está indicada la corrección quirúrgica: artrodesis. [ 9 ]
Se están realizando investigaciones tanto sobre el componente genético de la enfermedad como sobre sus métodos de tratamiento. El uso de células madre, ciertas hormonas, lecitina o ácido ascórbico aún no ha arrojado resultados positivos.
Pero gracias a investigaciones recientes, es posible que aparezca algo nuevo en el tratamiento de la enfermedad de Charcot-Marie-Tooth en un futuro próximo. Así, desde 2014, la empresa francesa Pharnext ha estado desarrollando, y desde mediados de 2019, ensayos clínicos con el fármaco PXT3003 para el tratamiento de la CMT tipo 1 en adultos. Este fármaco suprime la expresión aumentada del gen PMP22, mejora la mielinización de los nervios periféricos y alivia los síntomas neuromusculares.
Especialistas de la empresa médica Sarepta Therapeutics (EE. UU.) trabajan en el desarrollo de una terapia génica para la enfermedad de Charcot-Marie-Tooth tipo 1. Esta terapia utilizará un virus adenoasociado (VAA) inocuo del género Dependovirus con un genoma de ADN monocatenario lineal, que transferirá al organismo el gen NTF3, que codifica la proteína neurotrofina-3 (NT-3), necesaria para el funcionamiento de las células nerviosas de Schwann.
Helixmith comenzará los ensayos clínicos de la terapia génica Engensis (VM202), desarrollada en Corea del Sur, a finales de 2020 para tratar los síntomas musculares en la enfermedad de CMT tipo 1. [ 10 ]
Prevención
La prevención de la CMT puede consistir en el asesoramiento genético de los futuros padres, especialmente si algún miembro de la pareja tiene antecedentes familiares de la enfermedad. Sin embargo, se han identificado casos de mutaciones puntuales de genes de novo, es decir, sin antecedentes familiares de la enfermedad.
Durante el embarazo, la probabilidad de enfermedad de Charcot-Marie-Tooth en el futuro niño se puede comprobar mediante una biopsia de vellosidades coriónicas (entre las 10 y 13 semanas de gestación), así como con un análisis del líquido amniótico (entre las 15 y 18 semanas).
Pronóstico
En general, el pronóstico de los diferentes tipos de enfermedad de Charcot-Marie-Tooth depende de la gravedad clínica, pero en todos los casos la enfermedad progresa lentamente. Muchos pacientes presentan discapacidades, aunque esto no reduce la esperanza de vida.