Médico experto del artículo.

Nuevos artículos
Nefritis hereditaria (síndrome de Alport) en niños
Último revisado: 05.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
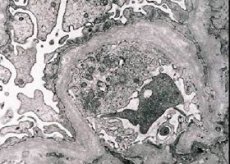
La nefritis hereditaria (síndrome de Alport) es una glomerulopatía no inmunológica hereditaria determinada genéticamente, que se manifiesta por hematuria (a veces con proteinuria), deterioro progresivo de la función renal con desarrollo de insuficiencia renal crónica, a menudo combinada con sordera neurosensorial y discapacidad visual.
La enfermedad fue descrita por primera vez en 1902 por LG Guthrie, quien observó una familia con hematuria en varias generaciones. En 1915, AF Hurst describió el desarrollo de uremia en miembros de la misma familia. En 1927, A. Alport identificó por primera vez la pérdida auditiva en varios familiares con hematuria. En la década de 1950, se describieron lesiones oculares en una enfermedad similar. En 1972, en pacientes con hematuria hereditaria, durante un estudio morfológico del tejido renal, Hinglais et al. revelaron una expansión y estratificación desiguales de las membranas basales glomerulares. En 1985, se identificó la base genética de la nefritis hereditaria: una mutación en el gen del colágeno tipo IV (Fiengold et al., 1985).
El estudio de la naturaleza genética de la enfermedad nos permitió concluir que las diferencias en las manifestaciones fenotípicas de la nefritis hereditaria (con o sin pérdida auditiva) se deben al grado de expresión del gen mutante. Por lo tanto, actualmente, todas las variantes clínicas se consideran manifestaciones de una misma enfermedad, y el término «nefritis hereditaria» es sinónimo de «síndrome de Alport».
Según estudios epidemiológicos, la nefritis hereditaria se presenta con una frecuencia de 17 por cada 100.000 niños.
 [
[ Causas del síndrome de Alport
La base genética de la enfermedad es una mutación en el gen de la cadena α-5 del colágeno tipo IV. Este tipo es común en las membranas basales del riñón, el aparato coclear, la cápsula del cristalino, la retina y la córnea, lo cual se ha demostrado en estudios con anticuerpos monoclonales contra esta fracción de colágeno. Recientemente, se ha sugerido la posibilidad de utilizar sondas de ADN para el diagnóstico prenatal de la nefritis hereditaria.
Se enfatiza la importancia de realizar pruebas de ADN a todos los miembros de la familia para identificar portadores del gen mutante, lo cual es fundamental para el asesoramiento médico y genético de familias con esta enfermedad. Sin embargo, hasta un 20% de las familias no tienen familiares con enfermedad renal, lo que sugiere una alta frecuencia de mutaciones espontáneas del gen anormal. La mayoría de los pacientes con nefritis hereditaria tienen familiares con enfermedad renal, pérdida auditiva y patología visual; los matrimonios consanguíneos entre personas con uno o más antepasados son importantes, ya que en el matrimonio entre individuos emparentados aumenta la probabilidad de recibir los mismos genes de ambos progenitores. Se han establecido vías de transmisión autosómica dominante, autosómica recesiva y dominante ligada al cromosoma X.
En los niños, se distinguen con mayor frecuencia tres tipos de nefritis hereditaria: síndrome de Alport, nefritis hereditaria sin pérdida auditiva y hematuria benigna familiar.
El síndrome de Alport es una nefritis hereditaria con pérdida auditiva. Se basa en un defecto combinado en la estructura del colágeno de la membrana basal glomerular de los riñones, el oído y los ojos. El gen del síndrome de Alport clásico se localiza en el locus 21-22 q del brazo largo del cromosoma X. En la mayoría de los casos, se hereda de forma dominante, ligado al cromosoma X. En este sentido, el síndrome de Alport es más grave en hombres, ya que en las mujeres la función del gen mutante se compensa con un alelo sano del segundo cromosoma intacto.
La base genética del desarrollo de la nefritis hereditaria reside en mutaciones en los genes de las cadenas alfa del colágeno tipo IV. Se conocen seis cadenas alfa del colágeno tipo IV G: los genes de las cadenas a5 y a6 (Col4A5 y Col4A5) se localizan en el brazo largo del cromosoma X, en la zona 21-22q; los genes de las cadenas a3 y a4 (Col4A3 y Col4A4) se localizan en el cromosoma 2; y los genes de las cadenas a1 y a2 (Col4A1 y Col4A2) se localizan en el cromosoma 13.
En la mayoría de los casos (80-85%), se detecta un patrón de herencia ligado al cromosoma X, asociado con daño en el gen Col4A5 como resultado de deleciones, mutaciones puntuales o trastornos de empalme. Actualmente, se han identificado más de 200 mutaciones del gen Col4A5, responsables de la interrupción de la síntesis de las cadenas α5 del colágeno tipo IV. Con este tipo de herencia, la enfermedad se manifiesta en niños de ambos sexos, pero en los varones es más grave.
Las mutaciones en los loci de los genes Col4A3 y Col4A4, responsables de la síntesis de las cadenas a3 y a4 del colágeno tipo IV, se heredan de forma autosómica. Según estudios, la herencia autosómica dominante se observa en el 16 % de los casos de nefritis hereditaria, y la autosómica recesiva, en el 6 % de los pacientes. Se conocen alrededor de 10 variantes de mutaciones en los genes Col4A3 y Col4A4.
El resultado de las mutaciones es una alteración de los procesos de ensamblaje del colágeno tipo IV, lo que conlleva una alteración de su estructura. El colágeno tipo IV es uno de los principales componentes de la membrana basal glomerular, el aparato coclear y el cristalino, cuya patología se detecta en la clínica de la nefritis hereditaria.
El colágeno tipo IV, que forma parte de la membrana basal glomerular, consta principalmente de dos cadenas α1 (IV) y una cadena α2 (IV), además de las cadenas α3, α4 y α5. Con mayor frecuencia, en la herencia ligada al cromosoma X, la mutación del gen Col4A5 se acompaña de la ausencia de las cadenas α3, α4, α5 y α6 en la estructura del colágeno tipo IV, y aumenta el número de cadenas α1 y α2 en la membrana basal glomerular. El mecanismo de este fenómeno no está claro; se asume que la causa son cambios postranscripcionales en el ARNm.
La ausencia de las cadenas a3, a4 y a5 en la estructura del colágeno tipo IV de las membranas basales glomerulares provoca su adelgazamiento y fragilidad en las primeras etapas del síndrome de Alport, que se manifiesta clínicamente con mayor frecuencia por hematuria (con menor frecuencia por hematuria con proteinuria o solo proteinuria), hipoacusia y lenticono. La progresión de la enfermedad provoca engrosamiento y deterioro de la permeabilidad de las membranas basales en las últimas etapas, con proliferación de colágeno tipo V y VI en ellas, lo que se manifiesta por un aumento de la proteinuria y una disminución de la función renal.
La naturaleza de la mutación subyacente a la nefritis hereditaria determina en gran medida su manifestación fenotípica. En caso de deleción del cromosoma X con mutación simultánea de los genes Col4A5 y Col4A6, responsables de la síntesis de las cadenas α5 y α6 del colágeno tipo IV, el síndrome de Alport se combina con leiomiomatosis esofágica y genital. Según datos de investigación, en caso de una mutación del gen Col4A5 asociada a una deleción, se observa una mayor gravedad del proceso patológico, una combinación de daño renal con manifestaciones extrarrenales y desarrollo temprano de insuficiencia renal crónica, en comparación con una mutación puntual de este gen.
Morfológicamente, la microscopía electrónica revela adelgazamiento y estratificación de las membranas basales glomerulares (especialmente la lámina densa) y la presencia de gránulos electrodensos. Las lesiones glomerulares pueden ser heterogéneas en un mismo paciente, desde lesiones mesangiales focales mínimas hasta glomeruloesclerosis. La glomerulitis en el síndrome de Alport siempre es inmunonegativa, lo que la distingue de la glomerulonefritis. Los rasgos característicos incluyen el desarrollo de atrofia tubular, infiltración linfohistiocítica y la presencia de células espumosas con inclusiones lipídicas (lipófagos). A medida que la enfermedad progresa, se revela un engrosamiento y una destrucción pronunciada de las membranas basales glomerulares.
Se revelan ciertos cambios en el sistema inmunitario. Los pacientes con nefritis hereditaria presentan niveles bajos de IgA y una tendencia a un aumento de la concentración de IgM en sangre. El nivel de IgG puede aumentar en las primeras etapas de la enfermedad y disminuir en las etapas posteriores. Quizás el aumento de la concentración de IgM y G sea una reacción compensatoria ante la deficiencia de IgA.
La actividad funcional del sistema de linfocitos T se reduce, se observa una disminución selectiva de los linfocitos B responsables de la síntesis de Ig A, se altera el vínculo fagocítico de la inmunidad, principalmente debido a la interrupción de los procesos de quimiotaxis y digestión intracelular en los neutrófilos.
Al examinar una biopsia renal en pacientes con síndrome de Alport, los datos de microscopía electrónica revelan cambios ultraestructurales en la membrana basal glomerular: adelgazamiento, alteración de la estructura y desdoblamiento de la membrana basal glomerular, con cambios en su grosor y contornos irregulares. En las primeras etapas de la nefritis hereditaria, este defecto determina el adelgazamiento y la fragilidad de la membrana basal glomerular.
El adelgazamiento de las membranas glomerulares es un signo más favorable y es más común en niñas. Un signo microscópico electrónico más constante en la nefritis hereditaria es la ruptura de la membrana basal, y la gravedad de su destrucción se correlaciona con la gravedad del proceso.
Síntomas del síndrome de Alport en niños
Los primeros síntomas del síndrome de Alport, en forma de síndrome urinario aislado, se detectan con mayor frecuencia en niños de los primeros tres años de vida. En la mayoría de los casos, la enfermedad se detecta por casualidad. El síndrome urinario se detecta durante una revisión preventiva del niño, antes de su ingreso en una guardería o durante una infección viral respiratoria aguda (IRV). En caso de patología en la orina durante una IVR, la nefritis hereditaria, a diferencia de la glomerulonefritis adquirida, no tiene período de latencia.
En la etapa inicial de la enfermedad, la salud del niño se ve levemente afectada; un rasgo característico es la persistencia y resistencia del síndrome urinario. Uno de los principales signos es la hematuria de diversa gravedad, observada en el 100% de los casos. Se observa un aumento de la hematuria durante o después de infecciones respiratorias, actividad física o vacunaciones preventivas. La proteinuria en la mayoría de los casos no supera 1 g/día; al inicio de la enfermedad puede ser inestable, pero a medida que progresa el proceso, aumenta. Periódicamente, puede presentarse leucocituria con predominio de linfocitos en el sedimento urinario, lo que se asocia con el desarrollo de cambios intersticiales.
Posteriormente, la función renal se deteriora parcialmente y el estado general del paciente empeora: aparecen intoxicación, debilidad muscular, hipotensión arterial, a menudo pérdida auditiva (especialmente en niños) y, en ocasiones, pérdida visual. La intoxicación se manifiesta con palidez, fatiga y cefalea. En la etapa inicial de la enfermedad, la pérdida auditiva se detecta, en la mayoría de los casos, solo mediante audiografía. La pérdida auditiva en el síndrome de Alport puede presentarse en diferentes períodos de la infancia, pero con mayor frecuencia se diagnostica entre los 6 y los 10 años. La pérdida auditiva en niños comienza con las frecuencias altas, alcanzando un grado significativo en la conducción aérea y ósea, pasando de la pérdida auditiva de conducción del sonido a la pérdida auditiva de percepción del sonido. La pérdida auditiva puede ser uno de los primeros síntomas de la enfermedad y preceder al síndrome urinario.
En el 20% de los casos, los pacientes con síndrome de Alport presentan cambios en los órganos visuales. Las anomalías detectadas con mayor frecuencia son las del cristalino: esferofoquia, lenticono anterior, posterior o mixto y diversas cataratas. En familias con síndrome de Alport, hay una frecuencia significativa de miopía. Varios investigadores notan constantemente cambios perimaculares bilaterales en estas familias en forma de granulaciones blanquecinas o amarillentas brillantes en el cuerpo lúteo. Consideran este signo como un síntoma constante que tiene un alto valor diagnóstico en el síndrome de Alport. KS Chugh et al. (1993) en un estudio oftalmológico encontraron en pacientes con síndrome de Alport una disminución de la agudeza visual en el 66,7% de los casos, lenticono anterior en el 37,8%, manchas retinianas en el 22,2%, cataratas en el 20% y queratocono en el 6,7%.
En algunos niños con nefritis hereditaria, especialmente cuando se desarrolla insuficiencia renal, se observa un retraso significativo en el desarrollo físico. A medida que la insuficiencia renal progresa, se desarrolla hipertensión arterial. En niños, se detecta con mayor frecuencia en la adolescencia y en edades más avanzadas.
Los pacientes con nefritis hereditaria se caracterizan por la presencia de varios (más de 5-7) estigmas de dismorfogénesis del tejido conectivo. Entre los estigmas del tejido conectivo, los más comunes son el hipertelorismo ocular, el paladar alto, las anomalías de la mordida, la forma anormal de las aurículas, la curvatura del meñique en las manos y la "sandalia" en los pies. La nefritis hereditaria se caracteriza por la uniformidad de los estigmas de dismorfogénesis dentro de una familia, así como por una alta frecuencia de su distribución entre los familiares de los probandos a través de cuya línea se transmite la enfermedad.
En las primeras etapas de la enfermedad, se detecta una disminución aislada de las funciones renales parciales: transporte de aminoácidos, electrolitos, función de concentración y acidogénesis. Los cambios posteriores afectan el estado funcional de las porciones proximal y distal de la nefrona y se caracterizan por trastornos parciales combinados. La disminución de la filtración glomerular se produce más tarde, con mayor frecuencia en la adolescencia. A medida que la nefritis hereditaria progresa, se desarrolla anemia.
Así, la nefritis hereditaria se caracteriza por una evolución escalonada de la enfermedad: primero, una fase latente o síntomas clínicos ocultos, que se manifiestan por cambios mínimos en el síndrome urinario; posteriormente, se produce una descompensación gradual del proceso con disminución de la función renal y síntomas clínicos evidentes (intoxicación, astenia, retraso del desarrollo, anemia). Los síntomas clínicos suelen aparecer independientemente de la estratificación de la reacción inflamatoria.
La nefritis hereditaria puede manifestarse en diferentes períodos de edad, lo que depende de la acción del gen, que se encuentra en estado reprimido hasta un momento determinado.
Clasificación
Hay tres tipos de nefritis hereditaria
- Opción I: se manifiesta clínicamente como nefritis con hematuria, hipoacusia y daño ocular. La nefritis evoluciona progresivamente con el desarrollo de insuficiencia renal crónica. El tipo de herencia es dominante, ligado al cromosoma X. Morfológicamente, se observa una alteración de la estructura de la membrana basal, su adelgazamiento y división.
- Opción II: se manifiesta clínicamente como nefritis con hematuria sin hipoacusia. La nefritis evoluciona progresivamente con el desarrollo de insuficiencia renal crónica. El tipo de herencia es dominante, ligado al cromosoma X. Morfológicamente, se detecta adelgazamiento de la membrana basal capilar glomerular (especialmente la laminadensa).
- Opción III: hematuria familiar benigna. La evolución es favorable y no se desarrolla insuficiencia renal crónica. El tipo de herencia es autosómico dominante o autosómico recesivo. En el caso del tipo autosómico recesivo, la enfermedad se presenta con mayor gravedad en mujeres.
Diagnóstico del síndrome de Alport
Se proponen los siguientes criterios:
- la presencia de al menos dos pacientes con nefropatía en cada familia;
- hematuria como síntoma principal de nefropatía en el probando;
- la presencia de pérdida auditiva en al menos un miembro de la familia;
- desarrollo de insuficiencia renal crónica en uno o más familiares.
En el diagnóstico de diversas enfermedades hereditarias y congénitas, se concede gran importancia a un enfoque integral del examen y, sobre todo, a la atención a los datos obtenidos durante la elaboración del árbol genealógico del niño. El diagnóstico del síndrome de Alport se considera válido cuando se detectan tres de los cuatro signos típicos en el paciente: presencia de hematuria e insuficiencia renal crónica en la familia, hipoacusia neurosensorial, patología visual y detección de signos de clivaje de la membrana basal glomerular con cambios en su grosor y contornos irregulares en la microscopía electrónica de la biopsia.
El examen del paciente debe incluir métodos de investigación clínica y genética; estudio específico del historial de la enfermedad; examen general del paciente teniendo en cuenta criterios diagnósticos significativos. En la etapa de compensación, la patología solo puede detectarse centrándose en síndromes como la presencia de una carga hereditaria, hipotensión, múltiples estigmas de disembriogénesis y cambios en el síndrome urinario. En la etapa de descompensación, pueden aparecer síntomas extrarrenales, como intoxicación grave, astenia, retraso en el desarrollo físico y anemia, que se manifiestan e intensifican con una disminución gradual de la función renal. En la mayoría de los pacientes, con una disminución de la función renal, se observa lo siguiente: disminución de la acidogénesis y la aminogénesis; el 50 % de los pacientes nota una disminución significativa de la función secretora de los riñones; rango limitado de fluctuaciones en la densidad óptica de la orina; alteración del ritmo de filtración y, posteriormente, una disminución de la filtración glomerular. La etapa de insuficiencia renal crónica se diagnostica cuando los pacientes tienen un nivel elevado de urea en el suero sanguíneo (más de 0,35 g/l) durante 3-6 meses o más, y una disminución de la filtración glomerular al 25% de la norma.
El diagnóstico diferencial de la nefritis hereditaria debe realizarse principalmente con la forma hematúrica de la glomerulonefritis adquirida. La glomerulonefritis adquirida suele tener un inicio agudo, en un período de 2 a 3 semanas después de la infección, con signos extrarrenales que incluyen hipertensión desde los primeros días (en la nefritis hereditaria, por el contrario, hipotensión), disminución de la filtración glomerular al inicio de la enfermedad y ausencia de deterioro de las funciones tubulares parciales, mientras que en la hereditaria sí lo están. La glomerulonefritis adquirida se presenta con hematuria y proteinuria más pronunciadas, con un aumento de la VSG. Los cambios típicos en la membrana basal glomerular, característicos de la nefritis hereditaria, tienen valor diagnóstico.
El diagnóstico diferencial de la nefropatía dismetabólica se realiza con la insuficiencia renal crónica y, en el caso de enfermedades renales heterogéneas clínicamente diagnosticadas en la familia, puede existir un espectro de nefropatía que va desde pielonefritis hasta urolitiasis. Los niños suelen quejarse de dolor abdominal y dolor periódico al orinar, con presencia de oxalatos en el sedimento urinario.
Si se sospecha nefritis hereditaria, el paciente debe ser remitido a un servicio de nefrología especializado para aclarar el diagnóstico.
¿Qué es necesario examinar?
Cómo examinar?
¿Qué pruebas son necesarias?
¿A quién contactar?
Tratamiento del síndrome de Alport
El régimen incluye restricciones en el esfuerzo físico intenso y la exposición al aire libre. La dieta es completa, con niveles adecuados de proteínas, grasas y carbohidratos, teniendo en cuenta la función renal. Es fundamental la detección y el tratamiento de focos infecciosos crónicos. Se utilizan los siguientes medicamentos: ATP, cocarboxilasa, piridoxina (hasta 50 mg/día) y cloruro de carnitina. Los tratamientos se administran de 2 a 3 veces al año. Para la hematuria, se prescriben hierbas medicinales: ortiga, jugo de aronia y milenrama.
Existen informes en la literatura nacional e internacional sobre el tratamiento con prednisolona y el uso de citostáticos. Sin embargo, es difícil evaluar su efecto.
En la insuficiencia renal crónica se utiliza la hemodiálisis y el trasplante de riñón.
No existen métodos de tratamiento patogénico específico y eficaz para la nefritis hereditaria. Todas las medidas terapéuticas están dirigidas a prevenir y ralentizar el deterioro de la función renal.
La dieta debe ser equilibrada y rica en calorías, considerando el estado funcional de los riñones. En ausencia de trastornos funcionales, la dieta del niño debe contener suficientes proteínas, grasas y carbohidratos. Si existen signos de disfunción renal, se debe limitar la cantidad de proteínas, carbohidratos, calcio y fósforo, lo que retrasa el desarrollo de insuficiencia renal crónica.
La actividad física debe ser limitada; se aconseja a los niños evitar los deportes.
Se debe evitar el contacto con pacientes infecciosos y reducir el riesgo de desarrollar enfermedades respiratorias agudas. Es necesario el saneamiento de los focos de infección crónica. No se administran vacunas preventivas a niños con nefritis hereditaria; la vacunación solo es posible por indicaciones epidemiológicas.
La terapia hormonal e inmunosupresora en la nefritis hereditaria es ineficaz. Existen indicios de cierto efecto positivo (reducción de la proteinuria y ralentización de la progresión de la enfermedad) con el uso prolongado de ciclosporina A e inhibidores de la ECA durante varios años.
En el tratamiento de los pacientes se utilizan medicamentos que mejoran el metabolismo:
- piridoxina - 2-3 mg/kg/día en 3 dosis durante 4 semanas;
- cocarboxilasa - 50 mg por vía intramuscular cada dos días, un total de 10-15 inyecciones;
- ATP - 1 ml por vía intramuscular cada dos días, 10-15 inyecciones;
- vitamina A - 1000 UI/año/día en 1 dosis durante 2 semanas;
- Vitamina E – 1 mg/kg/día en 1 dosis durante 2 semanas.
Este tipo de terapia ayuda a mejorar el estado general de los pacientes, reducir disfunciones tubulares y se realiza en cursos 3 veces al año.
El levamisol se puede utilizar como inmunomodulador: 2 mg/kg/día 2-3 veces por semana con intervalos de 3-4 días entre dosis.
Según datos de investigación, la oxigenación hiperbárica tiene un efecto positivo sobre la gravedad de la hematuria y la disfunción renal.
El método más eficaz para tratar la nefritis hereditaria es el trasplante renal oportuno. En este caso, no hay recaída de la enfermedad en el riñón trasplantado; en un pequeño porcentaje de casos (alrededor del 5%), puede desarrollarse nefritis en el riñón trasplantado asociada a antígenos de la membrana basal glomerular.
Una dirección prometedora es el diagnóstico prenatal y la terapia de ingeniería genética. Experimentos con animales muestran una alta eficiencia en la transferencia de genes normales responsables de la síntesis de cadenas alfa de colágeno tipo IV al tejido renal, tras lo cual se observa la síntesis de estructuras de colágeno normales.
Pronóstico
El pronóstico de la nefritis hereditaria es siempre grave.
Los criterios pronósticos desfavorables para la evolución de la nefritis hereditaria son:
- género masculino;
- desarrollo temprano de insuficiencia renal crónica en miembros de la familia;
- proteinuria (más de 1 g/día);
- engrosamiento de las membranas basales glomerulares según microscopía;
- neuritis acústica;
- deleción en el gen Col4A5.
El pronóstico de la hematuria familiar benigna es más favorable.
Использованная литература