Médico experto del artículo.

Nuevos artículos
Acondroplasia
Último revisado: 12.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
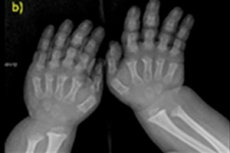
Existen muchas enfermedades congénitas raras, y una de ellas es la violación del crecimiento óseo: la acondroplasia, que conduce a una estatura baja desproporcionada grave.
En la sección sobre anomalías del desarrollo de la CIE-10, el código para este tipo de displasia osteocondral hereditaria con defectos de crecimiento de los huesos tubulares y la columna vertebral es Q77.4 [ 1 ]
Epidemiología
En cuanto a la prevalencia de la acondroplasia, los datos estadísticos de diversos estudios son ambiguos. Algunos afirman que esta anomalía se presenta en un recién nacido de cada 10 000, otros, en uno de cada 26 000 a 28 000, y otros, en entre 4 y 15 casos de cada 100 000. [ 2 ]
También existe información de que cuando el padre tiene más de 50 años, la incidencia de acondroplasia en los hijos es de un caso por cada 1875 recién nacidos.
Causas acondroplasia
La causa de la acondroplasia es una alteración de la osteogénesis, en particular, uno de los tipos de osificación intrauterina de las diáfisis de los huesos tubulares del esqueleto: la osificación endocondral, durante la cual el cartílago se transforma en tejido óseo. Para más detalles, consulte " Desarrollo y crecimiento óseo".
La alteración de la osificación de los huesos largos, es decir, la acondroplasia fetal, se produce debido a mutaciones en el gen de la tirosina quinasa de membrana, el receptor 3 del factor de crecimiento de fibroblastos (FGFR3 en el cromosoma 4p16.3), que afecta el crecimiento y la diferenciación celular. La presencia de mutaciones en el FGFR3 se asocia con inestabilidad genética y cambios en el número de cromosomas (aneuploidía).
La acondroplasia se transmite a un hijo como rasgo autosómico dominante, es decir, recibe una copia del gen mutante (dominante) y un gen normal en un par de cromosomas autosómicos. Por lo tanto, el tipo de herencia de este defecto es autosómico dominante, y la anomalía puede manifestarse en el 50 % de la descendencia cuando se cruza una combinación de alelos de este gen (genotipo).
Además, las mutaciones pueden ser esporádicas y, como demuestra la práctica, en el 80% de los casos los niños con acondroplasia nacen de padres de estatura normal.
Factores de riesgo
Los principales factores de riesgo para el nacimiento de niños con acondroplasia son hereditarios. Si uno de los padres presenta este defecto, la probabilidad de tener un hijo enfermo se estima en un 50 %; si ambos padres presentan esta anomalía, la probabilidad también es del 50 %, pero con un 25 % de riesgo de acondroplasia homocigótica, lo que puede provocar la muerte prenatal o en la primera infancia.
Con la edad del padre (cerca de los 40 años o más), aumenta el riesgo de una nueva mutación (mutación de novo) del gen FGFR3.
Patogenesia
Al explicar la patogenia de la acondroplasia, los expertos destacan la importancia de la proteína transmembrana tirosina proteína quinasa (codificada por el gen FGFR3) en la regulación de la división, diferenciación y apoptosis de las células del tejido cartilaginoso de las placas de crecimiento - condrocitos, así como el desarrollo normal del esqueleto - osteogénesis y mineralización del tejido óseo.
Durante el desarrollo embrionario, en presencia de una mutación genética, los receptores del factor de crecimiento de fibroblastos 3 se vuelven más activos. El aumento de sus funciones interrumpe la transmisión de señales celulares y la interacción de la parte extracelular de esta proteína con los factores de crecimiento de fibroblastos (FGF) polipeptídicos. Como resultado, se produce una falla: la etapa de proliferación de las células del cartílago se acorta y su diferenciación comienza antes de lo esperado. Todo esto conduce a una formación y fusión inadecuadas de los huesos del cráneo y a una displasia esquelética (disminución de los huesos largos, que se acompaña de baja estatura pronunciada o enanismo).
Y dos tercios de los casos de enanismo están asociados con acondroplasia.
Síntomas acondroplasia
El crecimiento óseo anormal provoca síntomas clínicos de acondroplasia como:
- estatura baja pronunciada (enanismo desproporcionado) con una altura adulta promedio de 123-134 cm;
- acortamiento de las partes proximales de los miembros inferiores y superiores con un tamaño de torso relativamente normal;
- dedos de manos y pies acortados;
- cabeza agrandada (macro o megalocefalia); [ 3 ]
- rasgos faciales específicos en forma de frente prominente e hipoplasia de la parte media de la cara (puente nasal hundido).
- Unión craneocervical estrecha. Algunos bebés con acondroplasia mueren durante el primer año de vida por complicaciones relacionadas con la unión craneocervical; estudios poblacionales sugieren que este riesgo de muerte puede alcanzar el 7,5 % sin evaluación e intervención.[ 4 ]
- La disfunción del oído medio suele ser un problema [ 5 ] y, si no se trata adecuadamente, puede provocar una pérdida auditiva conductiva lo suficientemente grave como para interferir con el desarrollo del habla. Más de la mitad de los niños necesitarán un tubo de ecualización de presión [ 6 ]. En general, alrededor del 40 % de las personas con acondroplasia presentan una pérdida auditiva funcionalmente significativa. El desarrollo del lenguaje expresivo también suele retrasarse, aunque la relación entre la pérdida auditiva y los problemas del lenguaje expresivo es cuestionable.
- La arqueación de la tibia es muy común en personas con acondroplasia. Más del 90 % de los adultos no tratados presentan algún grado de arqueación.[ 7 ] La "arqueación" es, en realidad, una deformidad compleja resultante de una combinación de inclinación lateral, torsión interna de la tibia e inestabilidad dinámica de la rodilla.[ 8 ]
Los bebés con acondroplasia se caracterizan por hipotonía muscular, lo que les permite aprender habilidades de movimiento y a caminar más tarde. Este defecto del desarrollo no afecta la inteligencia ni las capacidades cognitivas. [ 9 ], [ 10 ]
Consecuencias y complicaciones
Este tipo de displasia osteocondral hereditaria se caracteriza por las siguientes complicaciones y consecuencias:
- infecciones de oído recurrentes;
- apnea obstructiva del sueño;
- hidrocefalia;
- Maloclusión y dientes torcidos:
- deformación de las piernas (varo o valgo) con cambio en la marcha;
- lordosis hipertrofiada de la columna lumbar o su curvatura (cifosis toracolumbar o escoliosis lumbar) - con dolor de espalda al caminar;
- dolor en las articulaciones (debido a una posición incorrecta de los huesos o a la compresión de las raíces nerviosas);
- Estenosis espinal y compresión de la médula espinal; La queja médica más común en la edad adulta es la estenosis espinal sintomática que afecta L1-L4. Los síntomas varían desde claudicación intermitente y reversible inducida por el ejercicio hasta disfunción grave e irreversible de las piernas y retención urinaria.[ 11 ] La claudicación y la estenosis pueden causar síntomas tanto sensitivos (entumecimiento, dolor, pesadez) como motores (debilidad, tropiezos, resistencia limitada para caminar). La claudicación vascular resulta de la inflamación de los vasos sanguíneos después de estar de pie y caminar, y es completamente reversible con el reposo. La estenosis espinal es la lesión real de la médula espinal o la raíz nerviosa por el hueso estenótico del canal espinal, y los síntomas son irreversibles. Los síntomas localizados en un dermatoma particular pueden resultar de la estenosis de agujeros específicos de la raíz nerviosa.
- Reducción de la pared torácica con crecimiento pulmonar limitado y disminución de la función pulmonar (dificultad respiratoria grave). En la infancia, un pequeño grupo de personas con acondroplasia presenta problemas pulmonares restrictivos. La combinación de senos pequeños y mayor distensibilidad torácica resulta en una capacidad pulmonar reducida y enfermedad pulmonar restrictiva [ 12 ].
Otros problemas ortopédicos
- Debilidad articular. La mayoría de las articulaciones presentan hipermovilidad en la infancia. Generalmente, esto tiene poco efecto, salvo por la inestabilidad de la rodilla en algunas personas.
- Menisco lateral discoide: esta anomalía estructural recientemente identificada puede provocar dolor crónico de rodilla en algunas personas.[ 13 ]
- Artritis: La activación constitutiva de FGFR-3, como en la acondroplasia, puede proteger contra el desarrollo de la artritis.[ 14 ]
- La acantosis nigricans se observa en aproximadamente el 10% de las personas con acondroplasia.[ 15 ] En esta población, no refleja hiperinsulinemia ni malignidad.
La acondroplasia homocigótica causada por variantes patogénicas bialélicas en el nucleótido 1138 del FGFR3 es un trastorno grave con hallazgos radiológicos cualitativamente diferentes a los observados en la acondroplasia. La muerte prematura se debe a insuficiencia respiratoria debido a una pared torácica estrecha y a déficits neurológicos debidos a la estenosis cervicomedular [Hall, 1988].
Diagnostico acondroplasia
En la mayoría de los pacientes, el diagnóstico de acondroplasia se basa en los signos clínicos característicos y los hallazgos radiográficos. En lactantes o en ausencia de algunos síntomas, se utilizan pruebas genéticas, como el análisis del cariotipo, para establecer un diagnóstico definitivo.[16 ]
Al realizar el diagnóstico prenatal mediante el método de genética molecular, se pueden realizar análisis de líquido amniótico o de una muestra de vellosidades coriónicas.
Los signos de acondroplasia en la ecografía del feto (acortamiento de las extremidades y rasgos faciales típicos) se visualizan después de las 22 semanas de embarazo.
El diagnóstico instrumental también incluye radiografías del esqueleto o ecografías óseas. La radiografía confirma el diagnóstico basándose en datos como un cráneo grande con un agujero occipital estrecho y una base relativamente pequeña; huesos tubulares cortos y costillas acortadas; cuerpos vertebrales cortos y aplanados; conducto raquídeo estrecho y tamaño reducido de las alas ilíacas.
Diagnóstico diferencial
Es necesario el diagnóstico diferencial con enanismo hipofisario, displasia espondiloepifisaria congénita y diastrófica, hipocondroplasia, síndromes de Shereshevsky-Turner y de Noonan, y pseudoacondroplasia. Por lo tanto, la diferencia entre pseudoacondroplasia y acondroplasia radica en que, en pacientes con enanismo y pseudoacondroplasia, el tamaño de la cabeza y los rasgos faciales son normales.
¿A quién contactar?
Tratamiento acondroplasia
El Comité de Genética de la Academia Americana de Pediatría ha elaborado recomendaciones para el cuidado de niños con acondroplasia. Estas recomendaciones tienen como objetivo orientar y no sustituir la toma de decisiones individual. Una revisión reciente [Pauli y Botto, 2020] también incluye directrices. Existen clínicas especializadas en el tratamiento de la displasia esquelética; sus recomendaciones pueden diferir ligeramente de estas recomendaciones generales.
Las recomendaciones incluyen (pero no se limitan a) las siguientes.
Hidrocefalia. Si aparecen signos o síntomas de aumento de la presión intracraneal (p. ej., crecimiento acelerado de la cabeza, fontanela abultada persistente, dilatación notable de las venas superficiales faciales, irritabilidad, vómitos, alteraciones visuales, cefalea), es necesario derivar al paciente a un neurocirujano.
La etiología presunta de la hidrocefalia en la acondroplasia es el aumento de la presión venosa intracraneal debido a la estenosis de los agujeros yugulares. Por lo tanto, el tratamiento estándar ha sido la derivación ventriculoperitoneal. Sin embargo, la ventriculostomía endoscópica del tercer ventrículo puede ser beneficiosa en algunos individuos,[ 17 ] lo que implica que otros mecanismos, como la obstrucción del desfiladero del cuarto ventrículo debido a la estenosis craneocervical, podrían estar involucrados.[ 18 ]
Estenosis de la unión craneocervical. Mejores predictores de la necesidad de descompresión suboccipital:
- Hiperreflexia o clonus de las extremidades inferiores
- Hipopnea central en la polisomnografía
- Reducción del tamaño del foramen magnum determinada mediante tomografía computarizada de la unión craneocervical y comparada con las normas para niños con acondroplasia.[ 19 ]
- Recientemente se ha sugerido que la evidencia de compresión de la médula espinal y/o anomalías de la señal ponderada en T2 son otro factor a tener en cuenta al momento de decidir operar.
Si hay signos claros de compresión sintomática, se debe realizar una derivación urgente a un neurocirujano pediátrico para una cirugía de descompresión. [ 20 ]
El tratamiento de la apnea obstructiva del sueño puede incluir:
- Adenoamigdalectomía
- Presión positiva en las vías respiratorias
- Traqueotomía en casos extremos
- Pérdida de peso
Estas intervenciones pueden dar como resultado una mejora en los trastornos del sueño y cierta mejora en la función neurológica.[ 21 ]
En casos raros donde la obstrucción es lo suficientemente grave como para requerir una traqueotomía, se ha utilizado la cirugía de avance del tercio medio facial para aliviar la obstrucción de las vías respiratorias superiores.[ 22 ]
Disfunción del oído medio. Las infecciones frecuentes del oído medio, la acumulación persistente de líquido en el oído medio y la consiguiente pérdida auditiva deben tratarse de forma intensiva cuando sea necesario. Se recomienda el uso de tubos a largo plazo, ya que suelen ser necesarios hasta los siete u ocho años.[ 23 ]
Cuando surgen problemas a cualquier edad, se recomienda utilizar métodos de tratamiento adecuados.
Baja estatura. Diversos estudios han evaluado la terapia con hormona del crecimiento (GH) como posible tratamiento para la acondroplasia de baja estatura.[ 24 ]
En general, estas y otras series muestran una aceleración inicial del crecimiento, pero el efecto disminuye con el tiempo.
En promedio, se puede esperar un aumento en la altura adulta de solo unos 3 cm.
El alargamiento de extremidades mediante diversas técnicas sigue siendo una opción para algunas personas. Se pueden lograr aumentos de altura de hasta 30-35 cm. [ 25 ] Las complicaciones son frecuentes y pueden ser graves.
Si bien algunos recomiendan realizar estos procedimientos a los seis u ocho años de edad, muchos pediatras, genetistas clínicos y especialistas en ética recomiendan retrasar este tipo de cirugía hasta que el joven pueda participar en la toma de una decisión informada.
Al menos en Norteamérica, solo una pequeña proporción de las personas afectadas opta por someterse a un alargamiento avanzado de extremidades. El Consejo Asesor Médico de Little People of America ha emitido una declaración sobre el uso del alargamiento avanzado de extremidades.
Obesidad: Las medidas para prevenir la obesidad deben comenzar en la primera infancia. Los tratamientos estándar para la obesidad deberían ser eficaces en personas con acondroplasia, aunque las necesidades calóricas son menores. [ 26 ]
Se deben utilizar las tablas estándar de peso y peso para la talla específicas para la acondroplasia para el seguimiento del progreso. Es importante destacar que estas curvas no son perfectas; se obtuvieron a partir de miles de puntos de datos de personas con acondroplasia.
Los estándares del índice de masa corporal (IMC) se desarrollaron para niños de 16 años o menos. [ 27 ] El IMC no está estandarizado para adultos con acondroplasia; las comparaciones con las curvas de IMC para la altura promedio darán resultados engañosos. [ 28 ]
Deformidad en varo. Se recomienda un seguimiento ortopédico anual por parte de un profesional con experiencia en acondroplasia o un cirujano ortopédico. Se han publicado criterios para la intervención quirúrgica.[ 29 ]
La presencia de una curva sintomática progresiva requiere la derivación a un traumatólogo. La deformidad en varo asintomática no suele requerir corrección quirúrgica. Se pueden optar por diversas intervenciones (p. ej., crecimiento guiado con ocho placas, osteotomía en valgo y osteotomía desrotacional). No existen estudios controlados que comparen los resultados de las opciones de tratamiento.
Cifosis. Los bebés con acondroplasia suelen desarrollar cifosis flexible. Existe un protocolo para prevenir el desarrollo de cifosis angular fija, que incluye evitar cochecitos flexibles, columpios y portabebés. Se recomienda no sentarse sin apoyo; aplique siempre contrapresión en la espalda al sostener al bebé.
- La cifosis mejora significativamente o se resuelve en la mayoría de los niños después de adoptar una postura ortógrada y comenzar a caminar. [ 30 ]
- En los niños que no remiten espontáneamente después de aumentar la fuerza del tronco y comenzar a caminar, el uso de aparatos ortopédicos suele ser suficiente para prevenir la persistencia de la cifosis toracolumbar.[ 31 ]
- Si la cifosis grave persiste, puede ser necesaria una cirugía de columna para prevenir complicaciones neurológicas.[ 32 ]
Estenosis espinal: si se presentan signos y/o síntomas graves de estenosis espinal, es necesaria la derivación urgente a un especialista quirúrgico.
Generalmente se recomienda la laminectomía extendida y amplia. La relevancia del procedimiento depende del nivel (p. ej., torácico o lumbar) y del grado de estenosis. Los pacientes obtuvieron mejores resultados y mejor función cuanto antes se sometieron a la cirugía tras la aparición de los síntomas [ 33 ].
Vacunas: La acondroplasia no impide la vacunación rutinaria. Dado el mayor riesgo respiratorio, las vacunas DTaP, antineumocócica y antigripal son especialmente importantes.
Necesidades de adaptación: Debido a la baja estatura, podrían ser necesarias modificaciones del entorno. En la escuela, esto podría incluir taburetes, interruptores de luz más bajos, inodoros de altura adecuada u otros medios de accesibilidad, pupitres más bajos y reposapiés delante de las sillas. Todos los niños deberían poder salir del edificio de forma independiente en caso de emergencia. Las manos pequeñas y los tendones débiles pueden dificultar la motricidad fina. Las adaptaciones adecuadas incluyen el uso de un teclado más pequeño, bolígrafos con peso y superficies de escritura más lisas. La mayoría de los niños deberían tener un PEI o un plan 504.
Las extensiones de pedales casi siempre son necesarias para montar. También podrían requerirse modificaciones en la estación de trabajo, como mesas más bajas, teclados más pequeños, escalones y acceso al baño.
Socialización: Debido a la baja estatura muy notoria asociada con la acondroplasia, las personas afectadas y sus familias pueden tener dificultades para socializar y adaptarse a la escuela.
Los grupos de apoyo como Little People of America, Inc (LPA) pueden ayudar a las familias a abordar estos problemas a través del apoyo de pares, el ejemplo personal y programas de concienciación social.
La información sobre empleo, educación, derechos de las personas con discapacidad, adopción de niños de baja estatura, cuestiones médicas, vestimenta adecuada, dispositivos de adaptación y crianza de los hijos está disponible a través de un boletín nacional, seminarios y talleres.
No existe ningún medicamento ni tratamiento no farmacológico que pueda curar este defecto congénito.
La fisioterapia es el tratamiento más comúnmente utilizado; también puede ser necesario el tratamiento para la hidrocefalia (mediante derivación o ventriculostomía endoscópica), la obesidad, [ 34 ] la apnea, [ 35 ] la infección del oído medio o la estenosis espinal.
En algunas clínicas, cuando el niño cumple entre cinco y siete años, se realiza un tratamiento quirúrgico: alargamiento de los huesos de las espinillas, los muslos e incluso los huesos del hombro o corrección de la deformidad, con ayuda de operaciones y dispositivos ortopédicos especiales, en tres o cuatro etapas, cada una de las cuales dura hasta 6-12 meses.
Terapia bajo investigación
La administración de un análogo del péptido natriurético tipo C se encuentra en ensayos clínicos. Los resultados iniciales han demostrado su buena tolerancia y un aumento de la velocidad de crecimiento con respecto al valor basal en niños con acondroplasia ( centro del ensayo ). [ 36 ] El péptido natriurético tipo C conjugado también se encuentra actualmente en ensayos clínicos. [ 37 ] Otras consideraciones incluyen la inhibición de la tirosina quinasa [ 38 ], la meclizina [ 39 ] y un señuelo soluble recombinante de FGFR3 humano. [ 40 ]
Busque en clinicaltrials.gov en los EE. UU. y en el Registro de ensayos clínicos de la UE en Europa para obtener información sobre ensayos clínicos para una amplia gama de enfermedades y afecciones.
Prevención
La única medida preventiva es el diagnóstico prenatal de enfermedades congénitas. [ 41 ], [ 42 ]
Pronóstico
¿Cuánto tiempo viven las personas con acondroplasia? Unos 10 años menos que la esperanza de vida promedio.
Dado que los cambios patológicos en el tejido óseo y las articulaciones limitan el autocuidado y la movilidad, a los niños con este diagnóstico se les asigna la condición de discapacitados. A largo plazo, la mayoría de los pacientes tienen un pronóstico normal, pero con la edad, aumenta el riesgo de cardiopatía. [ 43 ]