Médico experto del artículo.

Nuevos artículos
Fibrosis quística
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
La fibrosis quística es una enfermedad genética autosómica recesiva monogénica caracterizada por un trastorno de la secreción de las glándulas exocrinas de órganos vitales con daño principalmente a los sistemas respiratorio y digestivo, de curso grave y pronóstico desfavorable.
 [ 1 ]
[ 1 ]
Epidemiología
La incidencia de la fibrosis quística fluctúa entre 1:2500 y 1:4600 recién nacidos. Cada año, nacen alrededor de 45 000 personas con fibrosis quística en todo el mundo. La incidencia de portadores del gen de la fibrosis quística es del 3-4 %, con aproximadamente 275 millones de personas en todo el mundo que son portadoras de este gen, de las cuales unos 5 millones viven en Rusia y unos 12,5 millones en los países de la CEI.
Causas fibrosis quística
La fibrosis quística se transmite de forma autosómica recesiva. El gen de la fibrosis quística se encuentra en el autosoma 7, contiene 27 exones y está compuesto por 250.000 pares de nucleótidos.
Un solo gen puede presentar numerosas mutaciones, cada una específica de una población o región geográfica. Se han descrito más de 520 mutaciones, la más común de las cuales es delta-P-508, es decir, una sustitución del aminoácido fenilalanina en la posición 508.
Patogenesia
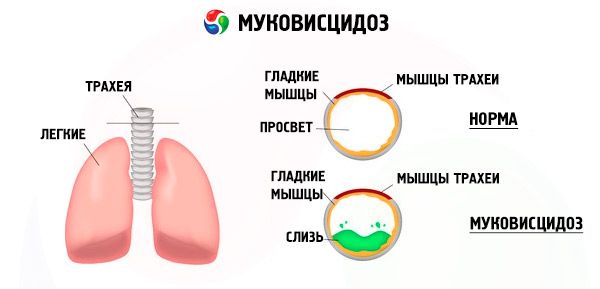
Las mutaciones en el gen de la fibrosis quística alteran la estructura y la función de la proteína CFTR (regulador transmembrana de la fibrosis quística). Esta proteína actúa como un canal de cloruro que participa en el intercambio hidroelectrolítico de las células epiteliales del sistema broncopulmonar, el tracto gastrointestinal, el páncreas, el hígado y el sistema reproductivo. Como resultado de la alteración de la función y la estructura de la proteína CFTR, los iones de cloruro Cl⁻ se acumulan en el interior de la célula. Esto provoca un cambio en el potencial eléctrico en el lumen de los conductos excretores, lo que facilita el flujo de grandes cantidades de iones de sodio (Na⁻ ) desde el lumen del conducto hacia la célula y mejora la absorción de agua del espacio pericelular.
Como resultado de estos cambios, la secreción de la mayoría de las glándulas exocrinas se espesa, su evacuación se altera, lo que conduce a trastornos secundarios pronunciados en órganos y sistemas, más pronunciados en los sistemas broncopulmonar y digestivo.
Se desarrolla un proceso inflamatorio crónico de intensidad variable en los bronquios. La función del epitelio ciliado se altera drásticamente, el esputo se vuelve muy viscoso, espeso y difícil de evacuar, se observa su estancamiento y se forman bronquiolo y bronquiectasias, que con el tiempo se vuelven más frecuentes. Estos cambios conducen a un aumento de la hipoxia y a la aparición de cardiopatía pulmonar crónica.
Los pacientes con fibrosis quística presentan una alta predisposición a desarrollar inflamación crónica en el sistema broncopulmonar. Esto se debe a alteraciones pronunciadas en el sistema de defensa broncopulmonar local (niveles reducidos de IgA, interferón y función fagocítica de los macrófagos alveolares y leucocitos).
Los macrófagos alveolares desempeñan un papel fundamental en el desarrollo de la inflamación crónica en el sistema broncopulmonar. Producen grandes cantidades de IL-8, lo que aumenta drásticamente la quimiotaxis de los neutrófilos en el árbol bronquial. Los neutrófilos se acumulan en grandes cantidades en los bronquios y, junto con las células epiteliales, secretan numerosas citocinas proinflamatorias, como IL-1, IL-8, IL-6, factor de necrosis tumoral y leucotrienos.
La alta actividad de la enzima elastasa también desempeña un papel importante en la patogénesis del daño del sistema broncopulmonar. Se distingue entre elastasa exógena y endógena. La primera es producida por la flora bacteriana (especialmente Pseudomonas aeruginosa), la segunda por los leucocitos neutrófilos. La elastasa destruye el epitelio y otros elementos estructurales de los bronquios, lo que contribuye a una mayor alteración del transporte mucociliar y a la rápida formación de bronquiectasias.
Los leucocitos neutrófilos también secretan otras enzimas proteolíticas. La alfa-1-antipirsina y el inhibidor secretor de leucoproteasas contrarrestan la influencia de las enzimas proteolíticas y, por lo tanto, protegen el sistema broncopulmonar de su efecto dañino. Sin embargo, lamentablemente, en pacientes con fibrosis quística, estos factores protectores se ven suprimidos por una cantidad significativa de proteasa neutrofílica.
Todas estas circunstancias contribuyen a la introducción de la infección en el sistema broncopulmonar y al desarrollo de bronquitis purulenta crónica. Además, debe tenerse en cuenta que la proteína defectuosa codificada por el gen de la fibrosis quística altera el estado funcional del epitelio bronquial, lo que favorece la adhesión de bacterias, principalmente Pseudomonas aeruginosa, a este epitelio.
Además de la patología del sistema broncopulmonar, la fibrosis quística también causa daños graves al páncreas, el estómago, el intestino grueso y delgado y el hígado.
Síntomas fibrosis quística
La fibrosis quística se manifiesta con diversos síntomas clínicos. En los recién nacidos, la enfermedad puede manifestarse con íleo meconial. Debido a la falta o incluso la ausencia total de tripsina, el meconio se vuelve muy denso y viscoso, acumulándose en la región ileocecal. Además, se desarrolla obstrucción intestinal, que se manifiesta con vómitos intensos con mezcla de bilis, distensión abdominal, falta de excreción de meconio, desarrollo de síntomas de peritonitis y un rápido aumento de las manifestaciones clínicas del síndrome de intoxicación grave. El niño puede morir en los primeros días de vida si no se realiza una intervención quirúrgica urgente.
En casos menos graves, un signo característico de la fibrosis quística son las heces abundantes y frecuentes, untuosas, con abundante grasa y de olor muy desagradable. En un tercio de los pacientes se observa prolapso rectal.
Posteriormente, los pacientes continúan experimentando disfunción intestinal, síndrome de malabsorción, trastornos graves del desarrollo físico e hipovitaminosis grave.
Durante el primer o segundo año de vida, aparecen síntomas de daño al sistema broncopulmonar (forma leve de la enfermedad), que se manifiestan con una tos que puede ser muy pronunciada y asemejarse a la tos ferina. La tos se acompaña de cianosis, disnea y expectoración espesa, inicialmente mucosa y posteriormente purulenta. Gradualmente, se desarrolla un cuadro clínico de bronquitis obstructiva crónica y bronquiectasias, enfisema pulmonar e insuficiencia respiratoria. Los niños son extremadamente susceptibles a las infecciones respiratorias agudas virales y bacterianas, lo que contribuye a las exacerbaciones y la progresión de la patología broncopulmonar. Es posible el desarrollo de asma bronquial dependiente de infecciones.
En niños en edad escolar, la fibrosis quística puede manifestarse como cólico intestinal. Los pacientes se quejan de dolor abdominal paroxístico intenso, distensión abdominal y vómitos repetidos. Al palpar el abdomen, se detectan formaciones densas en la proyección del intestino grueso: masas fecales mezcladas con moco espeso y denso. Los niños son muy propensos a desarrollar alcalosis hipoclorémica debido a la excreción excesiva de sal con el sudor en climas cálidos, lo que provoca la aparición de escarcha salina en la piel.
Trastornos del sistema broncopulmonar en adultos
El daño al sistema broncopulmonar en pacientes con fibrosis quística (forma pulmonar de la enfermedad) se caracteriza por el desarrollo de bronquitis obstructiva purulenta crónica, bronquiectasias, neumonía crónica, enfisema pulmonar, insuficiencia respiratoria y cardiopatía pulmonar. Algunos pacientes desarrollan neumotórax y otras complicaciones de la fibrosis quística: atelectasias, abscesos pulmonares, hemoptisis, hemorragia pulmonar y asma bronquial dependiente de infecciones.
Los pacientes se quejan de tos paroxística dolorosa con esputo mucopurulento muy viscoso y difícil de separar, a veces con sangre. Además, la disnea es muy característica, primero durante el esfuerzo físico y luego en reposo. La disnea se debe a una obstrucción bronquial. Muchos pacientes se quejan de rinitis crónica causada por poliposis y sinusitis. También son características la debilidad significativa, la disminución progresiva del rendimiento y las frecuentes enfermedades virales respiratorias agudas. En la exploración física, se observa palidez, hinchazón facial, cianosis de las mucosas visibles y disnea grave. Con el desarrollo de cardiopatía pulmonar descompensada, aparece edema en las piernas. Se puede observar engrosamiento de las falanges terminales de los dedos, en forma de baquetas, y uñas en forma de vidrios de reloj. El tórax adquiere forma de barril (debido al desarrollo de enfisema pulmonar).
La percusión pulmonar revela signos de enfisema: un ruido de caja, una marcada limitación de la movilidad del borde pulmonar y un descenso del borde inferior de los pulmones. La auscultación pulmonar revela respiración dificultosa con espiración prolongada, sibilancias secas dispersas y sibilancias húmedas, medianas y finas con burbujas. En el enfisema pulmonar grave, la respiración se ve gravemente debilitada.
Manifestaciones extrapulmonares de la fibrosis quística
Las manifestaciones extrapulmonares de la fibrosis quística pueden ser bastante pronunciadas y ocurrir con frecuencia.
Daño pancreático
En el 85% de los pacientes con fibrosis quística se observa insuficiencia pancreática exocrina de diversa gravedad. Con daño pancreático leve, no se presentan síndromes de malabsorción ni mala digestión; solo se presentan manifestaciones de laboratorio de insuficiencia exocrina (niveles bajos de tripsina y lipasa en sangre y contenido duodenal; a menudo, esteatorrea grave). Se sabe que para prevenir el síndrome de mala digestión, basta con la secreción de tan solo el 1-2% de la lipasa total. Solo se manifiestan clínicamente alteraciones significativas de la función exocrina.
En condiciones normales, los acinos del páncreas producen una secreción líquida rica en enzimas. A medida que la secreción se desplaza por el conducto excretor, se enriquece con agua y aniones, volviéndose aún más líquida. En la fibrosis quística, debido a un trastorno en la estructura y función del regulador transmembrana (canal de cloruro), la secreción pancreática no recibe suficiente líquido, se vuelve viscosa y su velocidad de movimiento por el conducto excretor disminuye drásticamente. Las proteínas de la secreción se depositan en las paredes de los conductos excretores pequeños, obstruyéndolos. A medida que la enfermedad progresa, se produce la destrucción y atrofia de los acinos, dando lugar a una pancreatitis crónica con insuficiencia pancreática exocrina. Esto se refleja clínicamente en el desarrollo de síndromes de malabsorción y mala digestión. La insuficiencia pancreática es la principal causa de malabsorción de grasas en la fibrosis quística, pero suele observarse con una deficiencia significativa de lipasa. Forsher y Durie (1991) indican que, en ausencia total de lipasa pancreática, la grasa se descompone y absorbe en un 50-60%, debido a la presencia de lipasas gástricas y salivales (sublinguales), cuya actividad se encuentra cerca del límite inferior de lo normal. Junto con la interrupción de la descomposición y absorción de las grasas, se produce una interrupción de la descomposición y reabsorción de las proteínas. Aproximadamente el 50% de la proteína ingerida con los alimentos se pierde en las heces. La absorción de carbohidratos se ve afectada en menor medida a pesar de la deficiencia de α-amilasa, pero el metabolismo de los carbohidratos puede verse significativamente afectado.
El daño al páncreas se expresa en el desarrollo del síndrome de mala digestión y malabsorción con pérdida de peso significativa y heces grasas abundantes.
El desarrollo de síndromes de mala digestión y malabsorción también se ve facilitado por una disfunción grave de las glándulas intestinales, una secreción alterada del jugo intestinal y una disminución del contenido de enzimas intestinales en él.
Los síndromes de mala digestión y malabsorción también se denominan forma intestinal de la fibrosis quística.
La alteración de la función endocrina del páncreas (diabetes mellitus) se observa en pacientes con fibrosis quística en las últimas etapas de la enfermedad (en el 2% de los niños y el 15% de los adultos).
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Daños en el hígado y las vías biliares
En el 13% de los pacientes con fibrosis quística mixta e intestinal, se desarrolla cirrosis hepática. Es más común en las mutaciones W128X, delta-P508 y X1303K. La cirrosis biliar hepática con hipertensión portal se detecta en el 5-10% de los pacientes. Según Welch, Smith (1995), se detectan signos clínicos, morfológicos, de laboratorio e instrumentales de daño hepático en el 86% de los pacientes con fibrosis quística.
Muchos pacientes con fibrosis quística también desarrollan colecistitis crónica, a menudo calculosa.
Disfunción de las glándulas sexuales
Los pacientes con fibrosis quística pueden experimentar azoospermia, causa de infertilidad. La fertilidad reducida también es común en las mujeres.
Etapa
Hay tres grados de gravedad de la fibrosis quística pulmonar.
La forma leve de la fibrosis quística se caracteriza por exacerbaciones raras (no más de una vez al año); durante los períodos de remisión, las manifestaciones clínicas están prácticamente ausentes y los pacientes pueden trabajar.
Gravedad moderada: las exacerbaciones se observan de 2 a 3 veces al año y duran aproximadamente 2 meses o más. En la fase de exacerbación, se presenta tos intensa con esputo difícil de separar, disnea incluso con un esfuerzo físico mínimo, fiebre baja, debilidad general y sudoración. Al mismo tiempo, se observa una alteración de la función exocrina del páncreas. En la fase de remisión, la capacidad laboral no se recupera por completo y persiste la disnea durante el esfuerzo físico.
El curso grave se caracteriza por exacerbaciones muy frecuentes de la enfermedad. Las remisiones son prácticamente inexistentes. En el cuadro clínico, destacan la insuficiencia respiratoria grave y los síntomas de cardiopatía pulmonar crónica, a menudo descompensada, con hemoptisis característica. Se observa una pérdida de peso significativa y los pacientes presentan una discapacidad completa. Por lo general, la patología broncopulmonar grave se acompaña de una disfunción pancreática grave.
Formas
- lesiones broncopulmonares
- Neumonía repetida y recurrente de curso prolongado.
- Neumonía abscesante, especialmente en lactantes.
- Neumonía crónica, especialmente bilateral.
- Asma bronquial refractaria al tratamiento tradicional.
- Bronquitis recurrente, bronquiolitis, especialmente con cultivo de Pseudomonas aeruginosa.
- Cambios en el tracto gastrointestinal
- Íleo meconial y sus equivalentes.
- Síndrome de absorción intestinal alterada de génesis desconocida.
- Ictericia obstructiva en recién nacidos de evolución prolongada.
- Cirrosis hepática.
- Diabetes mellitus.
- Reflujo gastroesofágico.
- Colelitiasis.
- Prolapso rectal.
- Cambios en otros órganos y sistemas
- Trastornos del crecimiento y del desarrollo.
- Desarrollo sexual retrasado.
- Infertilidad masculina.
- Pólipos nasales.
- Hermanos de familias con fibrosis quística.
[ 24 ]
Complicaciones y consecuencias
Las complicaciones del tracto gastrointestinal incluyen:
- La diabetes mellitus se desarrolla en el 8-12% de los pacientes mayores de 25 años.
- Colonopatía fibrosante.
- Íleo meconial en el período neonatal (en el 12% de los recién nacidos con fibrosis quística), síndrome de obstrucción intestinal distal, prolapso rectal, enfermedad ulcerosa péptica y enfermedad por reflujo gastroesofágico.
Complicaciones hepáticas:
- Enfermedad del hígado graso (en el 30-60% de los pacientes),
- Cirrosis biliar focal, cirrosis biliar multinodular e hipertensión portal asociada.
La hipertensión portal a veces conduce a la muerte debido a varices esofágicas.
La prevalencia de colecistitis y cálculos biliares es mayor en pacientes con fibrosis quística que en otros individuos.
Retraso de la pubertad, disminución de la fertilidad y otras complicaciones. La mayoría de los hombres presentan azoospermia y subdesarrollo de los conductos deferentes.
Diagnostico fibrosis quística
Análisis de sangre general: es típica la anemia de diversa gravedad, generalmente normo o hipocrómica. La anemia tiene una génesis polifactorial (reducción de la absorción de hierro y vitamina B12 en el intestino debido al desarrollo de síndrome de malabsorción). Es posible la leucopenia, con desarrollo de bronquitis purulenta y neumonía (leucocitosis y aumento de la VSG).
Análisis general de orina: sin cambios significativos, en casos raros se observa una ligera proteinuria.
Examen coprológico: se observa esteatorrea y creatrea. Becker (1987) recomienda medir la quimotripsina y los ácidos grasos en heces. Antes de determinar la quimotripsina en heces, es necesario suspender la toma de enzimas digestivas al menos 3 días antes del examen. En la fibrosis quística, la cantidad de quimotripsina en heces disminuye y la de ácidos grasos aumenta (la excreción normal de ácidos grasos es inferior a 20 mmol/día). Es importante tener en cuenta que también se observa una mayor excreción de ácidos grasos en las heces en:
- deficiencia de ácidos grasos conjugados en el intestino delgado debido a insuficiencia hepática, obstrucción de los conductos biliares, colonización bacteriana significativa del intestino delgado (en este caso, se produce una hidrólisis intensiva de los ácidos biliares);
- ileítis;
- enfermedad celíaca (con desarrollo del síndrome de malabsorción);
- enteritis;
- linfomas intestinales;
- enfermedad de Whipple;
- alergias alimentarias;
- tránsito acelerado de masas alimentarias en diarreas de diverso origen, síndrome carcinoide, tirotoxicosis.
Análisis de sangre bioquímico: disminución de los niveles de proteínas totales y albúmina, aumento de los niveles de alfa2 y gamma globulinas, bilirrubina y aminotransferasas (en caso de daño hepático), disminución de la actividad de la amilasa, lipasa, tripsina y niveles de hierro y calcio (en caso de desarrollo de síndrome de mala digestión, malabsorción).
Análisis de esputo: presencia de una gran cantidad de leucocitos neutrófilos y microorganismos (durante la bacterioscopia del esputo).
El estudio de la función de absorción del intestino delgado y de la función exocrina del páncreas revela alteraciones significativas.
Radiografía pulmonar: revela cambios cuya gravedad depende de la gravedad y la fase de la enfermedad. Los cambios más característicos son:
- aumento del patrón pulmonar debido a cambios intersticiales peribronquiales;
- expansión de las raíces de los pulmones;
- imagen de atelectasia lobulillar, subsegmentaria o incluso segmentaria de los pulmones;
- aumento de la transparencia de los campos pulmonares, principalmente en las secciones superiores, posición baja y movilidad insuficiente del diafragma, expansión del espacio retroesternal (manifestación de enfisema pulmonar);
- infiltración segmentaria o polisegmentaria del tejido pulmonar (en el desarrollo de neumonía).
La broncografía revela cambios provocados por la obstrucción de los bronquios por esputo viscoso (fragmentación del relleno de los bronquios con contraste, contornos desiguales, fenómeno de ruptura bronquial, disminución significativa del número de ramas laterales), así como broncoecgases (cilíndricos, mixtos), localizados principalmente en las partes inferiores de los pulmones.
La broncoscopia revela bronquitis purulenta difusa con esputo abundante, espeso y viscoso y películas fibrinosas.
Espirometría: ya en las primeras etapas de la enfermedad revela insuficiencia respiratoria de tipo obstructivo (disminución de FVC, FEV1, índice de Tiffno), restrictivo (disminución de FVC) o, más a menudo, obstructivo-restrictivo (disminución de FVC, FVC, FEV1, índice de Tiffno).
La prueba de sudor de Gibson y Cook (prueba de electrolitos en sudor) consiste en estimular la sudoración mediante electroforesis de pilocarpina, seguida de la determinación de cloruros en el sudor. Doerehuk (1987) describe la prueba de la siguiente manera: la electroforesis de pilocarpina se realiza en el antebrazo con una corriente eléctrica de 3 mA. Tras limpiar la piel con agua destilada, se recoge el sudor con un papel de filtro colocado sobre la zona estimulada y cubierto con una gasa para evitar la evaporación. Tras 30-60 minutos, se retira el papel de filtro y se diluye en agua destilada. Se mide la cantidad de sudor recogida. Para obtener resultados fiables, es necesario recoger al menos 50 mg (preferiblemente 100 mg) de sudor.
Si la concentración de cloruro es superior a 60 mmol/l, el diagnóstico de fibrosis quística se considera probable; si la concentración de cloruro es superior a 100 mmol/l, es fiable; en este caso, la diferencia en la concentración de cloro y sodio no debe superar los 8-10 mmol/l. Hadson (1983) recomienda que, si el contenido de sodio y cloruro en el sudor está en el límite, se realice una prueba de prednisolona (5 mg por vía oral durante 2 días, seguida de la determinación de electrolitos en el sudor). En personas que no padecen fibrosis quística, el nivel de sodio en el sudor disminuye hasta el límite inferior de lo normal; en la fibrosis quística, no cambia. Se recomienda una prueba del sudor para todo niño con tos crónica.
El análisis de muestras de sangre o ADN para detectar mutaciones importantes del gen de la fibrosis quística es la prueba diagnóstica más sensible y específica. Sin embargo, este método solo es adecuado para países donde la tasa de mutación delta-P508 es superior al 80 %. Además, la técnica es muy costosa y técnicamente compleja.
El diagnóstico prenatal de la fibrosis quística se realiza mediante la determinación de las isoenzimas de la fosfatasa alcalina en el líquido amniótico. Este método es posible entre las semanas 18 y 20 de embarazo.
Los principales criterios para el diagnóstico de la fibrosis quística son los siguientes:
- Indicaciones en la anamnesis de retraso del desarrollo físico en la infancia, enfermedades respiratorias crónicas recurrentes, trastornos dispépticos y diarrea, presencia de fibrosis quística en parientes cercanos;
- bronquitis obstructiva crónica, a menudo recurrente, con desarrollo de bronquiectasias y enfisema pulmonar, a menudo neumonía recurrente;
- pancreatitis crónica recurrente con marcada disminución de la función exocrina, síndrome de malabsorción;
- aumento del contenido de cloro en el sudor del paciente;
- Infertilidad con función sexual preservada.
El diagnóstico exitoso y el diagnóstico diferencial de la fibrosis quística se facilitan mediante la identificación de los grupos de riesgo.
Programa de detección de fibrosis quística
- Análisis general de sangre, orina, esputo.
- Análisis bacteriológico del esputo.
- Análisis coprológico.
- Análisis bioquímico de sangre: determinación de proteínas totales y fracciones proteicas, glucosa, bilirrubina, aminotransferasas, fosfatasa alcalina, gamma-glutamil transpeptidasas, potasio, calcio, hierro, lipasa, amilasa, tripsina.
- Estudio de la función exocrina del páncreas y de la función absortiva del intestino.
- Fluoroscopia y radiografía de pulmones, tomografía computarizada de pulmones.
- ECG.
- Ecocardiografía.
- Broncoscopia y broncografía.
- Espirometría.
- Prueba del sudor.
- Consulta con un genetista.
- Análisis de manchas de sangre o muestras de ADN para detectar mutaciones importantes del gen de la fibrosis quística.
¿Qué pruebas son necesarias?
¿A quién contactar?
Tratamiento fibrosis quística
El tipo y la gravedad de los síntomas de la fibrosis quística pueden variar mucho, por lo que no existe un plan de tratamiento típico; es individualizado para cada individuo.
La terapia consiste en las siguientes medidas terapéuticas:
- Los ejercicios de respiración y el drenaje postural ayudan a eliminar la mucosidad espesa que se acumula en los pulmones. Algunas técnicas para despejar las vías respiratorias requieren la ayuda de familiares, amigos o un neumólogo. Muchas personas usan un chaleco inflable para el pecho que vibra a alta frecuencia.

- Medicamentos inhalatorios que tienen efectos broncodilatadores, drenantes (mucolíticos) y antibacterianos (por ejemplo, fluoroquinolonas).
- Preparaciones que contienen enzimas pancreáticas para mejorar la digestión. Estas preparaciones se toman con las comidas.
- Multivitaminas (incluidas vitaminas liposolubles).
En 2015, la FDA aprobó un segundo fármaco para tratar la fibrosis quística, dirigido a una proteína defectuosa conocida como CFTR. El primer fármaco, un modulador del CFTR, se aprobó en 2012. Se espera que los moduladores del CFTR prolonguen décadas la vida de algunas personas con fibrosis quística.
Puede ser necesaria cirugía para tratar las siguientes complicaciones respiratorias:
- Neumotórax, hemoptisis masiva recurrente o persistente, pólipos nasales, sinusitis persistente y crónica.
- Íleo meconial, invaginación, prolapso rectal.
El trasplante de pulmón se realiza en la fase terminal de la enfermedad.
Pronóstico
La edad promedio de supervivencia de los pacientes con fibrosis quística oscila entre 35 y 40 años. Esta edad es mayor en hombres que en mujeres.
Con las estrategias de tratamiento modernas, el 80 % de los pacientes alcanza la edad adulta. Sin embargo, la fibrosis quística limita significativamente la capacidad funcional del paciente. Aún no existe cura para esta enfermedad.