Médico experto del artículo.

Nuevos artículos
Neuroblastoma en niños: causas, diagnóstico, tratamiento
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
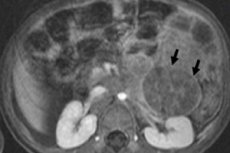
En oncología pediátrica, una de las neoplasias extracraneales más frecuentes es el neuroblastoma en niños, que es un tumor maligno embrionario de los neuroblastos de la cresta neural, es decir, células nerviosas embrionarias (inmaduras) del sistema nervioso simpático.
Epidemiología
Según las estadísticas del Grupo Internacional de Riesgo de Neuroblastoma (INRG), el neuroblastoma representa alrededor del 8% de todas las enfermedades oncológicas en niños en todo el mundo y ocupa el tercer lugar en prevalencia, después de la leucemia y los tumores cerebrales.
Según otros datos, el neuroblastoma representa aproximadamente el 28 % de todos los cánceres infantiles. Más de un tercio de los casos de neuroblastoma se diagnostican en niños menores de un año; la edad promedio de diagnóstico es de 19 a 22 meses. Más del 90 % de los casos diagnosticados se presentan en niños de dos a cinco años (con predominio de varones); la incidencia máxima se observa a los dos o tres años, y los casos en niños mayores de cinco años representan menos del 10 %.
Causas neuroblastomas
Al estudiar las causas del neuroblastoma, los investigadores concluyeron que este tumor infantil se produce debido a mutaciones genéticas esporádicas durante la embriogénesis o el desarrollo posnatal temprano. Sin embargo, se desconoce la causa de estos cambios genéticos, ya que no se ha identificado la influencia de factores ambientales teratogénicos.
Estos tumores pueden aparecer en cualquier parte, incluido el mediastino, el cuello, el abdomen, las glándulas suprarrenales, los riñones, la columna vertebral y la pelvis.
En casos raros, el neuroblastoma en bebés puede estar asociado a una mutación hereditaria. En particular, una mutación en el gen de la proteína de membrana CD246 (en el cromosoma 2), la enzima tirosina quinasa ALK, que garantiza la comunicación intercelular y desempeña un papel importante en el funcionamiento del sistema nervioso; y en el gen de la proteína PHOX2B (en el cromosoma 4), que participa en la maduración de las células nerviosas.
El neuroblastoma también puede estar asociado con la neurofibromatosis infantil tipo 1,el síndrome de Beckwith-Wiedemann y la hipoglucemia hiperinsulinémica (pancreatitis por nesidioblastosis).
Factores de riesgo
Hoy en día, la herencia se reconoce como un factor de riesgo para el desarrollo de neuroblastoma en niños: la presencia de este tumor en los antecedentes familiares, así como las anomalías congénitas asociadas a mutaciones genéticas durante el desarrollo intrauterino. Esto es especialmente cierto en los casos de desarrollo de varias neoplasias en diferentes órganos.
Los investigadores no han identificado ninguno de los factores exógenos que aumentan el riesgo de este tumor.
Patogenesia
El mecanismo de desarrollo de los neuroblastomas se debe a alteraciones en la diferenciación y maduración de las células de la cresta neural, líneas celulares bilaterales que se forman en los bordes del tubo neural a partir de la capa germinal ectodérmica del embrión humano. Estas células migran (se desplazan) y se diferencian en diversos tipos de células: neuronas sensoriales y autónomas, células neuroendocrinas y células de la médula suprarrenal, células del cartílago y los huesos craneofaciales, así como células pigmentarias.
En el neuroblastoma, los neuroblastos migrados no maduran, sino que continúan creciendo y dividiéndose, formando un tumor. La patogénesis de su formación está asociada a las siguientes mutaciones genéticas:
- con duplicación de parte de la secuencia cromosómica o duplicación de segmentos del gen LMO1 en el cromosoma 11, codificando la proteína RBTN1 en las células de la cresta neural del embrión;
- Con un cambio en el número de copias del gen NBPF10 en el cromosoma 1q21.1, que codifica la proteína DUF1220, responsable de la proliferación de células madre neurales humanas. Estos trastornos provocan la duplicación o la deleción de este cromosoma (la ausencia de una parte del ADN).
- con cambios en el gen supresor de tumores ATRX (en el cromosoma Xq21.1);
- Con la presencia de copias adicionales (amplificación) del gen del factor de transcripción N-Myc en el cromosoma 2, que codifica uno de los factores de transcripción (proteína de unión al ADN) que regula la actividad de otros genes y controla la proliferación de células precursoras durante la formación de proteínas para la formación de tejidos y órganos fetales. La amplificación de este gen lo convierte en un oncogén, lo que provoca una interrupción del ciclo celular, un aumento de la proliferación celular y la formación de tumores.
Síntomas neuroblastomas
Los primeros signos del neuroblastoma no son específicos y pueden incluir pérdida de apetito (y pérdida de peso), fatiga al alimentarse, fiebre y dolor en las articulaciones.
Los síntomas clínicos dependen de la localización del tumor primario y de la presencia de metástasis (que ocurren en el 60-73% de los casos).
Con mucha frecuencia, el neuroblastoma primario se localiza en la médula suprarrenal, que tiene un origen similar al de las células nerviosas. En niños menores de un año, el neuroblastoma suprarrenal se diagnostica en el 35-40% de los casos. Sus síntomas incluyen dolor abdominal, fiebre, pérdida de peso, dolor óseo, anemia o síndrome de Pepper concomitante: daño hepático difuso con hepatomegalia grave y síndrome de dificultad respiratoria.
El neuroblastoma retroperitoneal o neuroblastoma retroperitoneal en los niños, a medida que crece, comienza a presionar la vejiga o los intestinos, lo que puede causar problemas para orinar o defecar, hinchazón de las piernas (en los niños, el escroto se hincha).
El neuroblastoma del mediastino en niños (neuroblastoma mediastínico) suele presionar la vena cava superior, lo que puede causar hinchazón en la cara, el cuello, los brazos y la parte superior del tórax (con una coloración rojiza azulada y nódulos subcutáneos). Se presentan tos y sibilancias, dificultad para respirar (dificultad para respirar) o dificultad para tragar (disfagia); se observa inflamación de los ganglios linfáticos en el cuello, por encima de la clavícula y en las axilas.
La propagación de células tumorales a la médula ósea provoca anemia, trombocitopenia y leucopenia con tendencia al sangrado.
Con metástasis en la zona periorbitaria, aparecen ojeras o hematomas alrededor de los ojos. Este tumor también puede causar dolor de cabeza y mareos, exoftalmia (protrusión de los globos oculares) y, debido a la compresión de las terminaciones nerviosas, párpados caídos (ptosis) y disminución del tamaño de las pupilas (miosis).
El neuroblastoma abdominal o neuroblastoma de la cavidad abdominal en niños provoca la formación de sellos palpables en el abdomen, distensión abdominal, pérdida de apetito, estreñimiento y aumento de la presión arterial. Un tumor que presiona la médula espinal o una raíz nerviosa puede causar entumecimiento y debilidad en las extremidades, incapacidad para mantenerse de pie, gatear o caminar. Si los huesos se ven afectados, puede presentarse dolor óseo.
En caso de un tumor en estadio 3-4 en la cavidad abdominal con daño en los ganglios linfáticos, las células tumorales pueden ingresar al parénquima renal y luego se desarrolla un neuroblastoma extenso del riñón en los niños, lo que conduce a la interrupción de sus funciones.
Etapa
- El neuroblastoma en estadio 1 es un tumor primario que está localizado y aislado en un área del cuerpo; los ganglios linfáticos de ambos lados no están afectados.
- Neuroblastoma en estadio 2. En el estadio 2A, el tumor primario está confinado en una zona, pero es grande; no hay afectación de los ganglios linfáticos bilaterales. En el estadio 2B, los ganglios linfáticos del lado del cuerpo donde se encuentra el tumor presentan metástasis.
- Neuroblastoma estadio 3: el tumor primario cruza la médula espinal o la línea media del cuerpo, se encuentran metástasis unilaterales o bilaterales en los ganglios linfáticos.
- Neuroblastoma en estadio 4: el tumor se ha diseminado a ganglios linfáticos distantes, médula ósea, huesos, hígado u otros órganos. El estadio 4S se determina en niños menores de un año con un tumor primario localizado, con diseminación a la piel, el hígado o la médula ósea.
Sistema Internacional de Estadificación del Riesgo de Neuroblastoma (INRGSS)
El INRGSS utiliza factores de riesgo definidos por imágenes (IDRF), que son factores observados en pruebas de imágenes que pueden significar que un tumor será más difícil de extirpar.
El INRGSS divide los neuroblastomas en 4 estadios:
- L1: El tumor no se ha propagado desde su origen ni ha invadido estructuras vitales. Está limitado a una parte del cuerpo, como el cuello, el tórax o el abdomen.
- L2: El tumor no se ha propagado (hecho metástasis) lejos de donde comenzó (por ejemplo, puede haber crecido desde el lado izquierdo del abdomen hasta el lado izquierdo del pecho), pero tiene al menos un IDRF.
- M: El tumor ha hecho metástasis en una parte distante del cuerpo (excepto los tumores en la etapa EM).
- EM: Enfermedad metastásica en niños menores de 18 meses de edad, en la que el cáncer se ha diseminado únicamente a la piel, el hígado y/o la médula ósea.
Complicaciones y consecuencias
El neuroblastoma se caracteriza por complicaciones y consecuencias como:
- propagación (metástasis) a los ganglios linfáticos, la médula ósea, el hígado, la piel y los huesos;
- compresión de la médula espinal (que puede causar dolor y provocar parálisis);
- desarrollo del síndrome paraneoplásico (debido a la acción de ciertas sustancias químicas secretadas por el tumor, así como del antígeno disialogangliósido GD2 expresado por sus células), que se manifiesta por movimientos oculares involuntarios rápidos, alteración de la coordinación, calambres musculares y diarrea;
- recaídas después de finalizar la terapia primaria (como lo demuestra la práctica clínica, los neuroblastomas de alto riesgo tienen una recaída en el 50% de los casos).
Diagnostico neuroblastomas
El diagnóstico de neuroblastoma sospechado en un niño requiere examen, pruebas de laboratorio y estudios de imagen.
Se realizan análisis de sangre y orina para detectar catecolaminas (noradrenalina y dopamina) y ácidos homovanílico o vanililmandélico (formados durante el metabolismo de estas hormonas); un análisis de sangre para enolasa neuroespecífica, un ensayo inmunoabsorbente ligado a enzimas (ELISA) del suero sanguíneo y un análisis de médula ósea (una muestra se obtiene mediante punción aspirativa). Se realiza una prueba de ADN para determinar mutaciones y una biopsia para el examen citomorfológico del tejido tumoral.
Tras la toma de las muestras de biopsia, se envían a un laboratorio donde un patólogo (médico especializado en la identificación de células cancerosas) las examina al microscopio. Con frecuencia, también se realizan análisis de laboratorio especiales para determinar si el tumor es un neuroblastoma.
Si se trata de un neuroblastoma, las pruebas de laboratorio también pueden ayudar a determinar qué tan rápido podría crecer o propagarse el tumor, así como qué tratamientos podrían funcionar mejor.
El diagnóstico instrumental permite visualizar la neoplasia mediante ultrasonidos, rayos X, resonancia magnética o tomografía computarizada, PET con introducción de 18F-fluorodesoxiglucosa o gammagrafía con MIBG - gammagrafía con metayodobencilguanidina. [ 1 ]
Diagnóstico diferencial
El diagnóstico diferencial incluye ganglioneuroma benigno, ganglioneuroblastoma, rabdomiosarcoma y nefroblastoma.
¿A quién contactar?
Tratamiento neuroblastomas
En el neuroblastoma, el tratamiento depende del grupo de riesgo del paciente (estadio del proceso tumoral), la localización del tumor, las características genómicas de las células tumorales y la edad del niño. Puede incluir seguimiento, cirugía, quimioterapia, radioterapia, inmunoterapia y trasplante de células madre hematopoyéticas.
La quimioterapia neoadyuvante o adyuvante (preoperatoria o posoperatoria) para el neuroblastoma pediátrico, al igual que cualquier quimioterapia contra el cáncer, se administra en ciclos: el fármaco se administra durante varios días seguidos, seguidos de un descanso para que el cuerpo se recupere. Los ciclos suelen repetirse cada tres o cuatro semanas.
Se utilizan los siguientes medicamentos (y sus combinaciones): Ciclofosfamida, Cisplatino o Carboplatino, Doxorrubicina (Adriamicina), Vincristina, Etopósido.
Los efectos secundarios comunes de los medicamentos de quimioterapia incluyen caída del cabello, pérdida de apetito, fatiga, náuseas y vómitos, úlceras bucales, diarrea o estreñimiento. La quimioterapia puede dañar la médula ósea y causar una disminución del recuento de células sanguíneas.
La inmunoterapia dirigida (dirigida al antígeno tumoral GD2) utiliza fármacos del grupo de anticuerpos monoclonales (MAb anti-GD2) dinutuximab (Unituxin) y naxitamab. Se administran por vía intravenosa mediante infusión prolongada, en combinación con el factor estimulante de colonias de granulocitos y macrófagos (citocina GM-CSF) e interleucina-2.
Los efectos secundarios de estos medicamentos incluyen dolor (a menudo muy intenso), disminución de la presión arterial, aumento del ritmo cardíaco, dificultad para respirar (con posible hinchazón de las vías respiratorias), aumento de la temperatura, náuseas, vómitos y diarrea, cambios en la composición celular y mineral de la sangre.
Para reducir el riesgo de recurrencia del cáncer después de la quimioterapia de dosis alta y el trasplante de células madre, los niños con neuroblastoma de alto riesgo son tratados con retinoides sistémicos, ácido 13-cis-retinoico (isotretinoína). [ 2 ]
Tratamiento quirúrgico del neuroblastoma: extirpación del tumor, por ejemplo, adrenalectomía abierta o resección laparoscópica del neuroblastoma suprarrenal; linfectomía (extirpación de los ganglios linfáticos afectados), etc. [ 3 ]
En el caso del neuroblastoma de alto riesgo, se puede utilizar radioterapia.[4 ]
Prevención
Dadas las causas del neuroblastoma en niños, la única medida preventiva podría ser el asesoramiento genético al planificar un embarazo. Sin embargo, debe tenerse en cuenta que este tumor se asocia con mutaciones hereditarias solo en el 1-2% de los casos.
Pronóstico
El neuroblastoma infantil tiene la capacidad de regresar espontáneamente.
Marcadores pronósticos
- Los tumores de alto riesgo, así como el neuroblastoma en niños de todas las edades y en todos los estadios (excepto el estadio 4S) –con mayor expresión del gen N-MYC y amplificación del oncogén N-Myc– tienen un pronóstico desfavorable que afecta la esperanza de vida.
- Tener células tumorales con ciertas partes de los cromosomas 1 u 11 faltantes (deleciones 1p u 11q) conlleva un peor pronóstico. Tener una parte extra del cromosoma 17 (ganancia 17q) también se asocia con un peor pronóstico.
- Las células de neuroblastoma con gran cantidad de ADN tienen un mejor pronóstico, especialmente en niños menores de 2 años.
- Los neuroblastomas que tienen más receptores de neurotrofina, especialmente el receptor del factor de crecimiento nervioso TrkA, tienen un mejor pronóstico.
Supervivencia según el grupo de riesgo del Grupo de Oncología Infantil (COG)
- Grupo de bajo riesgo: Los niños del grupo de bajo riesgo tienen una tasa de supervivencia a 5 años superior al 95%.
- Grupo de riesgo intermedio: Los niños del grupo de riesgo intermedio tienen una tasa de supervivencia a 5 años del 90% al 95%.
- Grupo de alto riesgo: Los niños del grupo de alto riesgo tienen una tasa de supervivencia a 5 años de aproximadamente el 50%.
Aproximadamente el 15% de las muertes por cáncer infantil se deben al neuroblastoma. La probabilidad de supervivencia a largo plazo para esta neoplasia maligna de alto riesgo no supera el 40%. La tasa general de supervivencia a cinco años es del 67-74%, del 43% en el grupo de uno a cuatro años y superior al 80% para el neuroblastoma diagnosticado durante el primer año de vida.
Использованная литература