La enfermedad de von Willebrand es una deficiencia congénita del factor de von Willebrand (FVW) que provoca disfunción plaquetaria. Generalmente se caracteriza por sangrado leve. Las pruebas de detección muestran un tiempo de sangrado prolongado, recuentos plaquetarios normales y, posiblemente, un ligero aumento del tiempo de tromboplastina parcial.
La púrpura trombocitopénica trombótica y el síndrome hemolítico urémico son enfermedades agudas y fulminantes caracterizadas por el desarrollo de trombocitopenia y anemia hemolítica microangiopática.
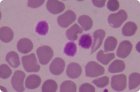
La púrpura trombocitopénica idiopática (inmunitaria) es un trastorno hemorrágico causado por trombocitopenia no asociada a una enfermedad sistémica. Suele ser crónica en adultos, pero a menudo es aguda y transitoria en niños. El bazo tiene un tamaño normal.
Los trastornos plaquetarios intracelulares hereditarios son enfermedades poco frecuentes que provocan hemorragias de por vida. El diagnóstico se confirma mediante pruebas de agregación plaquetaria. Si la hemorragia es grave, se requieren transfusiones de plaquetas.
Las disfunciones plaquetarias adquiridas pueden ser resultado de la aspirina, otros medicamentos antiinflamatorios no esteroides o enfermedades sistémicas.
Las plaquetas son fragmentos de megacariocitos que facilitan la hemostasia de la sangre circulante. La trombopoyetina es sintetizada por el hígado en respuesta a una disminución del número de megacariocitos y plaquetas circulantes en la médula ósea, y estimula la síntesis de plaquetas en la médula ósea a partir de los megacariocitos.
Las consecuencias de la linfopenia incluyen el desarrollo de infecciones oportunistas y un mayor riesgo de cáncer y enfermedades autoinmunes. Si se detecta linfopenia durante un hemograma completo, son necesarias pruebas diagnósticas para inmunodeficiencias y un análisis de la subpoblación linfocitaria. El tratamiento se centra en la enfermedad subyacente.
La neutropenia (agranulocitosis, granulocitopenia) es una disminución del número de neutrófilos (granulocitos) en la sangre. En casos graves, aumenta el riesgo y la gravedad de infecciones bacterianas y fúngicas.
Debido a la alta incidencia de talasemia HbS y beta talasemia en ciertos grupos de población, la presencia congénita de ambas anomalías es bastante común.
Los síntomas y molestias se deben al desarrollo de anemia, hemólisis, esplenomegalia, hiperplasia de médula ósea y, con múltiples transfusiones, sobrecarga de hierro. El diagnóstico se basa en el análisis cuantitativo de hemoglobina.