Médico experto del artículo.

Nuevos artículos
Priones: agentes causantes de las enfermedades priónicas
Último revisado: 06.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
Las infecciones virales lentas se caracterizan por criterios especiales:
- un período de incubación inusualmente largo (meses, años);
- una lesión específica de órganos y tejidos, principalmente del sistema nervioso central;
- progresión lenta y constante de la enfermedad;
- resultado fatal inevitable.

Algunos patógenos que causan infecciones virales agudas también pueden causar infecciones virales lentas. Por ejemplo, el virus del sarampión a veces causa pesquefiosis esclerosante subaguda (PEES), y el virus de la rubéola causa rubéola congénita progresiva y panencefalitis rubéola.
Una infección viral lenta típica en animales es causada por el virus visna/madi, un retrovirus. Es el agente causal de la infección viral lenta y la neumonía progresiva en ovejas. La sustancia blanca del cerebro se destruye, se desarrolla parálisis (visna, atrofia) y se produce inflamación crónica de los pulmones y el bazo.
Las enfermedades con características similares a las infecciones virales lentas son causadas por priones, los agentes causales de las infecciones priónicas. Las enfermedades priónicas son un grupo de trastornos progresivos del sistema nervioso central en humanos y animales. En los humanos, la función del sistema nervioso central se ve afectada, se producen cambios de personalidad y trastornos del movimiento. Los síntomas de la enfermedad suelen durar desde varios meses hasta varios años, y pueden resultar en la muerte. Anteriormente, las infecciones priónicas se consideraban junto con los llamados agentes causales de las infecciones virales lentas.
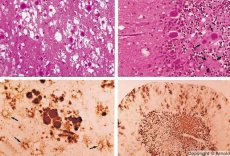
Algunos agentes causantes de enfermedades priónicas se acumulan primero en los tejidos linfoides. Al penetrar en el cerebro, los priones se acumulan en grandes cantidades, causando amiloidosis (disproteinosis extracelular, caracterizada por el depósito de amiloide con atrofia y esclerosis tisular) y astrocitosis (proliferación de neuroglia astrocítica e hiperproducción de fibras gliales). Se forman fibrillas, agregados de proteína o amiloide y cambios espongiformes en el cerebro (encefalopatías espongiformes transmisibles). Como resultado, se producen cambios en el comportamiento, alteración de la coordinación de movimientos y agotamiento con desenlace fatal. No se desarrolla inmunidad. Las enfermedades priónicas son enfermedades conformacionales que se desarrollan como resultado del plegamiento incorrecto (alteración de la conformación correcta) de las proteínas celulares necesarias para el funcionamiento normal del organismo. Las vías de transmisión priónica son diversas:
- Vía alimentaria - productos infectados de origen animal, aditivos alimentarios procedentes de órganos bovinos crudos, etc.:
- transmisión a través de transfusiones de sangre, administración de medicamentos de origen animal, trasplante de órganos y tejidos, uso de instrumentos quirúrgicos y dentales infectados;
- Transmisión a través de preparados inmunobiológicos (se conoce la infección de 1500 ovejas con PrP''' mediante vacuna de formol cerebral procedente de ovejas enfermas).
Los priones patológicos, tras entrar en el intestino, se transportan a la sangre y la linfa. Tras su replicación periférica en el bazo, el apéndice, las amígdalas y otros tejidos linfoides, se transfieren al cerebro a través de los nervios periféricos (neuroinvasión). Es posible la penetración directa de los priones en el cerebro a través de la barrera hematoencefálica. Anteriormente, se creía que el sistema nervioso central era el único tejido donde se acumulaban los priones patológicos, pero estudios han cambiado esta hipótesis. Se ha descubierto que la acumulación de priones en el bazo está asociada con el crecimiento y la función de las células dendríticas foliculares.
 [
[ Propiedades de los priones
La isoforma celular normal de la proteína priónica, con un peso molecular de 33-35 kDa, está determinada por el gen de la proteína priónica (el gen priónico, PrNP, se encuentra en el cromosoma 20). Este gen se encuentra en la superficie celular (anclado en la membrana por la glicoproteína de la molécula) y es sensible a la proteasa. Regula la transmisión de los impulsos nerviosos, los ciclos diarios y los procesos de oxidación, participa en el metabolismo del cobre en el sistema nervioso central y en la regulación de la división de las células madre de la médula ósea. Además, el gen priónico se encuentra en el bazo, los ganglios linfáticos, la piel, el tracto gastrointestinal y las células dendríticas foliculares.
Proliferación de priones patológicos
La transformación de los priones en formas alteradas ocurre cuando se altera el equilibrio cinético entre ellos. El proceso se ve potenciado por un aumento en la cantidad de priones patológicos (PrP) o exógenos. La PrP es una proteína normal anclada en la membrana celular. La PrP' es una proteína globular hidrofóbica que forma agregados consigo misma y con la PrP'' en la superficie celular: como resultado, la PrP' se transforma en PrP'' y el ciclo continúa. La forma patológica de la PrP''' se acumula en las neuronas, dando a la célula una apariencia esponjosa.
Kuru
Enfermedad priónica, anteriormente común entre los papúes (que significa temblor o sacudida) en la zona oriental de la isla de Nueva Guinea. Las propiedades infecciosas de la enfermedad fueron demostradas por K. Gajdusek. El patógeno se transmite por los alimentos como resultado del canibalismo ritual: ingerir el cerebro de familiares fallecidos, mal cocinado e infectado con priones. Como resultado del daño al sistema nervioso central, el movimiento y la marcha se ven afectados, y aparecen escalofríos y euforia ("muerte riendo"). El período de incubación dura de 5 a 30 años. El paciente fallece al año.
Enfermedad de Creutzfeldt-Jakob
Enfermedad priónica, que se manifiesta con demencia, trastornos visuales y cerebelosos, y trastornos del movimiento, con desenlace fatal tras 4-5 meses de enfermedad en la variante clásica de la enfermedad de Creutzfeldt-Jakob y tras 3-14 meses en la nueva variante. El periodo de incubación puede alcanzar los 20 años. Existen diversas vías de infección y causas posibles:
- al consumir productos animales insuficientemente tratados térmicamente, como carne y cerebros de vacas con encefalopatía espongiforme bovina;
- durante el trasplante de tejidos, como el trasplante de córnea, la transfusión de sangre, el uso de hormonas y otras sustancias biológicamente activas de origen animal, el uso de catgut, instrumentos quirúrgicos contaminados o insuficientemente esterilizados, manipulaciones prosectorales;
- en caso de hiperproducción de PrR y otras condiciones que estimulan el proceso de conversión de PrR' en PrR".
La enfermedad también puede desarrollarse como resultado de una mutación o inserción en la región del gen priónico. La naturaleza familiar de la enfermedad es común debido a la predisposición genética a la enfermedad de Creutzfeldt-Jakob. En la nueva variante de la enfermedad de Creutzfeldt-Jakob, los trastornos se desarrollan a una edad más temprana (edad promedio de 28 años), a diferencia de la variante clásica (edad promedio de 65 años). En la nueva variante de la enfermedad de Creutzfeldt-Jakob, la proteína priónica anormal se acumula no solo en el sistema nervioso central, sino también en los tejidos linforreticulares, incluidas las amígdalas.
Síndrome de Gerstmann-Sträussler-Scheinker
Enfermedad priónica hereditaria, acompañada de demencia, hipotonía, trastorno de la deglución (disfagia) y disartria. Suele tener un carácter familiar. El período de incubación es de 5 a 30 años. La enfermedad se presenta entre los 50 y 60 años y su duración oscila entre 5 y 13 años.
Insomnio fatal hereditario
Enfermedad autoinmune con insomnio progresivo, hiperreactividad simpática (hipertensión, hipertermia, hiperhidrosis, taquicardia), temblor, ataxia, multiclonación y alucinaciones. El sueño se ve gravemente afectado. La muerte se produce con la progresión de la insuficiencia cardiovascular.
Raspar
La tembladera (del inglés scrape - raspar) es una enfermedad priónica de las ovejas y las cabras (sarna), que cursa con daño al sistema nervioso central, trastornos progresivos del movimiento, picazón intensa de la piel (sarna) y termina en la muerte del animal.
Encefalopatía espongiforme bovina
Enfermedad del ganado que se caracteriza por daño al sistema nervioso central, alteración de la coordinación de movimientos y la muerte inevitable del animal. La epidemia se originó en Gran Bretaña. Se asoció con la alimentación de animales con harina de carne y hueso que contenía priones patógenos. El período de incubación oscila entre 1,5 y 15 años. El cerebro, la médula espinal y los globos oculares de los animales son los más infectados.
Diagnóstico de laboratorio de enfermedades priónicas
Durante el diagnóstico, se observan cambios espongiformes en el cerebro, astrocitosis (gliosis) y ausencia de infiltrados inflamatorios. Se tiñe el cerebro para amiloide. Se detectan marcadores proteicos de trastornos cerebrales priónicos en el líquido cefalorraquídeo (mediante ELISA). Se realiza un análisis genético del gen priónico (PCR).
Prevención de enfermedades priónicas
Para la descontaminación de instrumental y objetos ambientales se recomienda la esterilización en autoclave (a 134 °C durante 18 min; a 121 °C durante 1 h), la incineración y un tratamiento adicional con lejía y una solución de NaCl mononormal durante 1 h. Para la profilaxis no específica, se han restringido el uso de medicamentos de origen animal y se prohíbe la producción de hormonas hipofisarias de origen animal. El trasplante de duramadre está restringido. Se utilizan guantes de goma al manipular fluidos dialógicos de los pacientes.