Médico experto del artículo.

Nuevos artículos
Displasia renal
Último revisado: 04.07.2025

Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
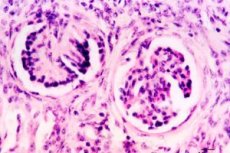
La displasia renal ocupa un lugar destacado entre los defectos del desarrollo del sistema urinario. Constituye un grupo heterogéneo de enfermedades asociadas con un deterioro del desarrollo del tejido renal. Morfológicamente, la displasia se basa en una diferenciación deficiente del blastema nefrogénico y las ramas del brote ureteral, con la presencia de estructuras embrionarias en forma de focos de mesénquima indiferenciado, así como conductos y túbulos primitivos. El mesénquima, representado por células cambiales pluripotentes y fibras de colágeno, puede formar derivados disontogenéticos del cartílago hialino y las fibras musculares lisas.
 [ 1 ]
[ 1 ]
Causas displasia renal
El papel principal en el desarrollo de la displasia renal lo desempeñan los trastornos genéticos (57%), la proporción de influencia teratogénica es significativamente menor (16%) y en casi un tercio de los pacientes se desconocen los factores que llevaron a la displasia.
Patogenesia
El examen morfológico de la displasia hipoplásica revela cierta disminución de la masa renal, una superficie lobulillar, una división en capas no siempre claramente definida y, en ocasiones, cierta expansión o hipoplasia de los uréteres. Microscópicamente, se detectan estructuras primitivas: muchos glomérulos presentan una reducción de tamaño, las asas vasculares están atróficas y la cápsula está engrosada. La forma de los glomérulos puede ser en S o anular, y muchos de ellos están hialinizados y esclerosados. Los glomérulos se disponen en forma de uva, rodeados de tejido conectivo laxo con acumulaciones focales de células linfoides e histiocíticas. En la médula, existen numerosos conductos y túbulos primitivos, que son formaciones inmaduras de diversas etapas del desarrollo embrionario. Los conductos primitivos se detectan principalmente en la zona yuxtamedular y son los restos del conducto mesonefrogénico. Un rasgo característico es la presencia de sombras de células musculares lisas y fibras de tejido conectivo a su alrededor. La presencia de estructuras primitivas refleja un retraso en la maduración de la nefrona.
El examen morfológico de la displasia focal simple no revela cambios significativos en la masa renal. En algunos casos, se observa una disminución del grosor de la corteza. Esta nefropatía se diagnostica basándose en los cambios histológicos revelados por microscopía. La displasia focal simple se caracteriza por la presencia de grupos de glomérulos y túbulos primitivos, rodeados de fibras de tejido conectivo y células musculares lisas, principalmente en la corteza renal; en ocasiones, se encuentra tejido cartilaginoso. El polimorfismo del epitelio de los túbulos contorneados es característico, donde las células adyacentes difieren en tamaño, configuración, disposición y número de orgánulos intracelulares. Algunos niños pueden presentar lúmenes tubulares dilatados en los riñones. También se pueden detectar quistes glomerulares, pero su número es insignificante. Se determinan células mononucleares mesenquimales en el estroma.
La displasia segmentaria simple (riñón Ask-Upmark) es bastante rara (0,02 % de todas las autopsias). En este tipo de displasia, el riñón presenta un tamaño reducido, se observa claramente un surco transversal en la superficie externa en el sitio del segmento hipoplásico y se reduce el número de pirámides. Los cambios morfológicos son causados por la disembriogénesis de vasos en segmentos individuales del riñón, con la consiguiente interrupción de la diferenciación de las estructuras tisulares debido a cambios en el suministro de sangre a estas áreas. Generalmente se detecta un subdesarrollo de las ramas arteriales correspondientes. Un rasgo característico es la presencia de conductos mesonefrogénicos primitivos en el segmento hipoplásico, rodeados de células musculares lisas y focos de cartílago hialino. Además, se desarrollan esclerosis, hialinosis glomerular, atrofia del epitelio tubular con expansión de su luz, signos de fibrosis e infiltración celular, e intersticio.
La displasia quística aplásica (riñón rudimentario multiquístico) representa el 3,5 % de todas las malformaciones congénitas del sistema urinario y el 19 % de todas las formas de displasia quística. Los riñones presentan un tamaño significativamente reducido, son formaciones quísticas sin forma de 2 a 5 mm de diámetro, el parénquima renal está casi completamente ausente, el uréter está ausente o presenta atresia. Microscópicamente, se detecta una gran cantidad de quistes, tanto glomerulares como tubulares, así como conductos primitivos y focos de tejido cartilaginoso. El daño bilateral es incompatible con la vida. El riñón rudimentario unilateral se detecta a menudo durante un examen aleatorio, y el segundo riñón suele ser anormal.
La displasia quística hipoplásica (riñón hipoplásico multiquístico) representa el 3,9 % de todos los defectos del sistema urinario y el 21,2 % de las displasias quísticas. Los riñones están reducidos en tamaño y peso. Los quistes glomerulares suelen localizarse en la zona subcapsular, su diámetro varía y puede alcanzar los 3-5 mm. Los quistes tubulares se encuentran tanto en la corteza como en la médula. La fibrosis del tejido conectivo y la presencia de conductos primitivos son más significativas en la médula. Los quistes son grandes y representan túbulos colectores dilatados quísticamente. El parénquima renal está parcialmente conservado. Entre las áreas patológicamente alteradas hay túbulos colectores de estructura normal. La pelvis renal puede permanecer sin cambios, más a menudo hipoplásica, al igual que el uréter. La displasia quística hipoplásica a menudo se asocia con defectos del tracto urinario inferior, el tracto gastrointestinal, el sistema cardiovascular y otros órganos.
El daño bilateral conduce tempranamente al desarrollo de insuficiencia renal crónica. Por lo general, en la variante unilateral de esta displasia, el segundo riñón presenta algunas manifestaciones de disembriogénesis.
La displasia quística hiperplásica suele acompañar al síndrome de Patau. El proceso es bilateral. Los riñones están agrandados y cubiertos de múltiples quistes. El examen microscópico revela conductos primitivos y numerosos quistes en la corteza y la médula. El desenlace fatal suele ocurrir a una edad temprana.
La displasia multiquística (riñón multiquístico) es un defecto del desarrollo en el que los riñones están agrandados en tamaño, hay una gran cantidad de quistes de diversas formas y tamaños (de 5 mm a 5 cm), entre los cuales el parénquima está prácticamente ausente.
La microscopía revela conductos primitivos y glomérulos entre los quistes, y también pueden encontrarse áreas con tejido cartilaginoso. En caso de lesiones bilaterales, la muerte ocurre en los primeros días de vida. En caso de lesiones unilaterales, el diagnóstico se realiza por casualidad durante la palpación de una formación tumoral tuberosa o basándose en los resultados de la ecografía. En caso de enfermedad multiquística unilateral, puede haber malformaciones en el segundo riñón (a menudo hidronefrosis), defectos cardíacos, defectos del tracto gastrointestinal, etc.
En la displasia medular (displasia quística de la médula, enfermedad quística medular, nefronoptisis de Fanconi), los riñones suelen presentar un tamaño reducido, conservando a menudo la lobulación embrionaria. La corteza se adelgaza y la médula se expande debido a la presencia de numerosos quistes de hasta 1 cm de diámetro, incluyendo la expansión quística característica de los túbulos colectores. La microscopía revela una disminución del tamaño de numerosos glomérulos; algunos de ellos están hialinizados y escleróticos; el intersticio también está esclerótico, y se observa infiltración linfoide en el estroma.
La enfermedad renal poliquística ocupa un lugar especial entre las displasias quísticas. Su aparición se asocia con una alteración del desarrollo embrionario renal, generalmente por una falta de conexión entre los túbulos colectores primarios y una parte de la nefrona derivada de un blastoma metanefrogénico. Los túbulos ciegos formados en este caso continúan desarrollándose, y la orina primaria se acumula en ellos, lo que los estira y provoca atrofia epitelial. Simultáneamente, crece el tejido conectivo que rodea los túbulos.
El tamaño de los quistes varía ampliamente: junto con los pequeños, visibles solo con una lupa o incluso un microscopio, los hay grandes, de hasta varios centímetros de diámetro. Una gran cantidad de quistes de paredes delgadas en la corteza y la médula de los riñones les da la apariencia de un panal al corte. Histológicamente, los quistes están representados por túbulos dilatados con epitelio cúbico o tienen la apariencia de cavidades con una pared gruesa de tejido conectivo y un epitelio marcadamente aplanado. E. Potter (1971) describió quistes asociados con la expansión de la cavidad de la cápsula de Bowman de los glomérulos, sin cambiar los túbulos. Los quistes pueden estar vacíos o contener líquido seroso, proteico, a veces teñido con pigmentos sanguíneos y cristales de ácido úrico. El estroma de los riñones en la enfermedad poliquística es esclerótico, a menudo con infiltración focal de células linfoides, y en niños menores de un año, con focos de hematopoyesis extramedular. A veces se encuentran islotes de cartílago o fibras musculares lisas en el estroma. El número y tipo de glomérulos y túbulos ubicados entre los quistes puede variar.
Síntomas displasia renal
La displasia total simple se describe a menudo en la literatura como displasia hipoplásica. Entre todas las malformaciones congénitas del sistema urinario, representa el 2,7%.
Se distingue entre variantes aplásicas e hipoplásicas. En el caso de la displasia renal aplásica, con lesiones bilaterales, la muerte se produce en las primeras horas o días de vida.
La variante hipoplásica se caracteriza por la manifestación temprana del síndrome urinario, caracterizado por mosaicismo, y el desarrollo temprano de insuficiencia renal crónica.
La displasia focal simple suele diagnosticarse mediante nefrobiopsia o autopsia. No presenta manifestaciones clínicas.
En la displasia segmentaria simple, el síntoma dominante es el desarrollo de hipertensión arterial persistente ya a una edad temprana, más frecuente en niñas. Los niños se quejan de cefaleas, pueden presentar convulsiones y se desarrollan precozmente alteraciones vasculares del fondo de ojo.
Uno de los principales síntomas clínicos es el síndrome doloroso, que se manifiesta en forma de dolor abdominal. La poliuria y la polidipsia aparecen bastante temprano como manifestaciones del síndrome tubulointersticial. En algunos casos, se observa un retraso en el peso corporal y el crecimiento de los niños. El síndrome urinario se manifiesta por proteinuria predominante en un contexto de microhematuria y leucocituria moderada.
Los signos clínicos de la enfermedad renal poliquística aparecen en la adolescencia: dolor lumbar, palpación de una formación tumoral en la cavidad abdominal e hipertensión arterial. El síndrome urinario se manifiesta con hematuria. A menudo se acompaña de pielonefritis. Funcionalmente, los riñones se conservan durante muchos años; posteriormente, aparecen hipostenuria, disminución de la filtración glomerular y azoemia.
El quiste multilocular (displasia quística focal del riñón) es una forma focal de displasia quística del riñón y se caracteriza por la presencia de un quiste multicámara en uno de sus polos, limitado por una cápsula de tejido renal normal y dividido internamente por septos.
El cuadro clínico de un quiste multilocular se caracteriza por la aparición de un síndrome doloroso de intensidad variable en el abdomen y la región lumbar, debido a la interrupción del flujo urinario por la compresión de la pelvis renal o el uréter por un quiste de gran tamaño. Además, debido a la posible compresión de los órganos abdominales, se presentan síntomas que simulan su enfermedad.
Las manifestaciones clínicas de la displasia medular generalmente se desarrollan después de alcanzar la edad de 3 años, más a menudo a la edad de 5-6 años aparece el "complejo de síntomas de Fanconi": poliuria, polidipsia, aumento de la temperatura corporal, retraso en el desarrollo físico, vómitos repetidos, deshidratación, acidosis, anemia, progresión rápida de la uremia.
El cuadro clínico de la displasia quística aplásica está determinado por el estado del segundo riñón, en el que a menudo se desarrolla pielonefritis debido a la presencia de displasia en él.
La displasia multiquística puede manifestarse con dolor sordo o paroxístico en el abdomen, así como en la región lumbar. Puede detectarse hipertensión arterial.
En la displasia cortical (enfermedad renal microquística, síndrome nefrótico congénito de tipo "finlandés"), los riñones no presentan cambios de tamaño y la lobulación puede conservarse. Se detectan pequeños quistes glomerulares y tubulares de 2 a 3 mm de diámetro. El síndrome nefrótico se observa desde el nacimiento. El síndrome nefrótico congénito de tipo "finlandés" es hormonorresistente y tiene un pronóstico desfavorable. Se observa el desarrollo temprano de insuficiencia renal crónica.
El cuadro clínico de la displasia quística hipoplásica es causado por pielonefritis, el desarrollo de insuficiencia renal crónica, cuya tasa de progresión depende no solo de la cantidad de parénquima conservado del riñón hipoplásico, sino también del grado de daño al segundo riñón no hipoplásico, pero, por regla general, que tiene elementos displásicos.
La displasia hipoplásica puede detectarse en el contexto de una enfermedad intercurrente, mientras que los síndromes extrarrenales pueden estar ausentes o manifestarse débilmente. El síndrome urinario se manifiesta por hematuria con proteinuria moderada. Las manifestaciones de esta enfermedad son muy heterogéneas. A menudo puede presentarse una variante proteinúrica con pérdida significativa de proteínas, pero el síndrome edematoso es relativamente raro, incluso con proteinuria significativa, y el síndrome nefrótico se caracteriza por ser incompleto. La observación dinámica del niño muestra que el cuadro clínico se caracteriza posteriormente por síndrome nefrótico, la presencia de cambios tubulointersticiales, a menudo con la estratificación de una infección del tracto urinario.
Los niños con displasia hipoplásica suelen desarrollar estados de hipoinmune o inmunodeficiencia, lo que explica la aparición de enfermedades intercurrentes graves y frecuentes con la progresión del proceso patológico renal. Una característica importante de esta nefropatía es la ausencia de hipertensión arterial; la hipotensión es más frecuente. El aumento de la presión arterial se produce ya con el desarrollo de insuficiencia renal crónica.
El curso de la displasia hipoplásica es tórpido, no hay ciclicidad ni naturaleza ondulatoria de las manifestaciones y la terapia farmacológica suele ser ineficaz.
Formas
Actualmente, no existe una clasificación generalmente aceptada de la displasia renal. La mayoría de los autores, basándose en las manifestaciones morfológicas, distinguen entre displasias simples y quísticas, y por localización: cortical, medular y corticomedular. Según la prevalencia, se distinguen displasias focales, segmentarias y totales.
Dependiendo de la prevalencia, existen formas totales, focales y segmentarias de displasia quística.
Entre las formas totales de displasia quística se distinguen las variantes aplásica, hipoplásica, hiperplásica y multiquística.
La enfermedad poliquística se manifiesta en dos formas principales, que difieren en la naturaleza de la herencia, las manifestaciones clínicas y el cuadro morfológico: los tipos "infantil" y "adulto".
La enfermedad poliquística infantil (riñón quístico pequeño) se hereda de forma autosómica recesiva. Los riñones presentan un tamaño y peso significativamente mayores. Se observan numerosos quistes cilíndricos y fusiformes en la corteza y la médula. Estos quistes están delimitados por escasas capas de tejido conectivo. También se encuentran quistes en el hígado y otros órganos. Las manifestaciones clínicas dependen del número de túbulos afectados. Con daño en el 60% de los túbulos, la muerte por uremia progresiva ocurre en los primeros 6 meses. Los resultados de O.V. Chumakova (1999) no confirman los conceptos clásicos de mortalidad precoz en niños con enfermedad poliquística autosómica recesiva y muestran que su esperanza de vida puede ser bastante larga, incluso con la detección temprana de los síntomas clínicos. Sin embargo, la insuficiencia renal crónica se desarrolla antes en estos niños que en la forma autosómica dominante de la enfermedad poliquística. En estos pacientes, los síntomas de daño hepático son los principales responsables del cuadro clínico. En la clínica se observan con frecuencia microhematuria y macrohematuria, así como hipertensión arterial. La enfermedad poliquística suele complicarse con pielonefritis de evolución tórpida.
En la enfermedad poliquística del tipo "adulto" (riñón quístico grande), los riñones casi siempre presentan un tamaño aumentado; su masa en adultos alcanza 1,5 kg o más cada uno. En la corteza y la médula se encuentran numerosos quistes de hasta 4-5 cm de diámetro.
Diagnostico displasia renal
El diagnóstico de la enfermedad renal poliquística se basa en los antecedentes familiares, datos ecográficos, urografía excretora, que muestran un aumento de los contornos de los riñones, aplanamiento de la pelvis renal con elongación, alargamiento y compresión de los cálices.
En el diagnóstico de los quistes multiloculares, los métodos de examen radiológico, incluida la nefrotomografía y la angiografía, son de importancia decisiva.
Entre los signos de laboratorio de la displasia medular, la hipoproteinemia es característica. El síndrome urinario suele manifestarse con proteinuria leve. Debido al aumento de la pérdida de sales, se desarrollan hiponatremia, hipopotasemia e hipocalcemia. La acidosis se desarrolla debido a una bicarbonaturia significativa, una alteración de la acidogénesis y la amoniogénesis.
El diagnóstico de la displasia quística aplásica se basa en datos ecográficos, urografía excretora, reno y gammagrafía. Durante la cistoscopia, el orificio ureteral del lado del riñón rudimentario suele estar ausente o estenótico.
Para el diagnóstico de la displasia hipoplásica son de gran importancia la detección accidental de la enfermedad, la presencia de múltiples estigmas de disembriogénesis y cierto retraso en el desarrollo físico.
Cómo examinar?
¿Qué pruebas son necesarias?
Diagnóstico diferencial
Tratamiento displasia renal
El tratamiento de la displasia hipoplásica es sintomático.
Si se detecta enfermedad multiquística se realiza nefrectomía por el riesgo de desarrollar malignidad.
El tratamiento de la displasia medular es sintomático. En caso de insuficiencia renal crónica, se indica hemodiálisis o diálisis peritoneal y trasplante renal.
Pronóstico
El pronóstico de la displasia hipoplásica es grave, con desarrollo temprano de insuficiencia renal crónica y necesidad de terapia sustitutiva (hemodiálisis o diálisis peritoneal, trasplante de riñón).
[ 28 ]
Использованная литература